
Table of contents
Introduction
Data collection
Database organisation
Navigation
Text Search
BLAST search
Detailed view of an entry
Download
Introduction
Transmembrane proteins constitute approximately 20 to 30% of fully sequenced proteomes and are responsible for a wide variety of cellular functions. Knowledge of the structure of proteins is crucial to understanding their function. However, due to the difficulty of obtaining crystals of transmembrane proteins suitable for crystallographic analyses, biochemical and computational methods are routinely used to determine their topology. Biochemical methods include: techniques of gene fusions, using enzymes such as alkaline phosphatase, β-galactosidase, β-lactamase, and various fluorescent proteins, detection of post-translational modifications such as glycosylation, phosphorylation and biotinylation, cysteine-scanning mutagenesis, proteolysis methods and epitope mapping techniques. The information we acquire from such experiments is very important and can be combined with computational methods in order to produce more reliable topological models of proteins.
Return to top
Data collection
We performed an extensive literature search (up to January 2007), in order to find studies that reported the use of biochemical methods for the determination of the topology of transmembrane proteins. The information was evaluated manually and and the data are organized in a Mysql database using PHP for creating the web interface.
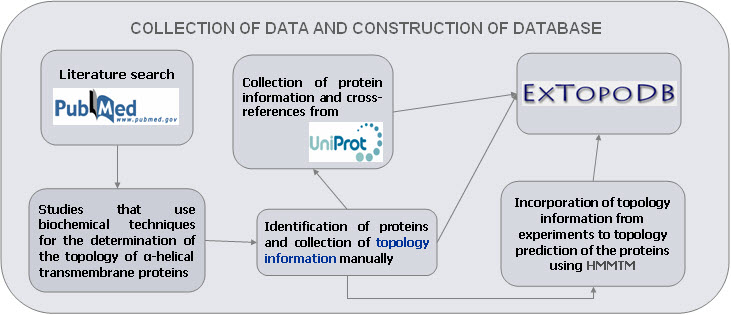
Figure 1. The methodology that was used for the collection of the data.
Return to top
Database organisation
ExTopoDB is a publicly accessible database of experimentally derived topological models of transmembrane proteins. It contains experimental information about the topology of 1354 transmembrane proteins. This information was collected from studies in the literature that reported the use of biochemical methods for the determination of the topology of transmembrane proteins. Biochemical methods include: techniques of gene fusions, using enzymes such as alkaline phosphatase, ß-galactosidase, ß-lactamase, and various fluorescent proteins, detection of post-translational modifications such as glycosylation, phosphorylation and biotinylation, cysteine-scanning mutagenesis, proteolysis methods and epitope mapping techniques. The information is provided with references to the literature. Each record contains information about the protein's sequence with cross-references to many publicly available databases worldwide. The user may submit advanced queries for text search and there is an interface for running BLAST against the database. Furthermore, the results of topology prediction using the HMM-TM algorithm are included for each protein in the database (unconstrained prediction) and we also incorporated the experimental information about the topology of the proteins, in the HMM-TM prediction procedure, producing more reliable topology models (constrained prediction). |
|
|
Return to top
Navigation
Through the navigation tool, the user has the ability to browse the database by organism or by the experimental method that was used for the determination of the topology of the protein entry.
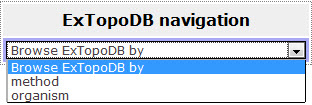
Figure 2. Basic view of the Navigation page. The user can choose to browse by experimental methods or by organisms.
If the user chooses to browse by Experimental methods he can choose from a drop-down list of biochemical methods.
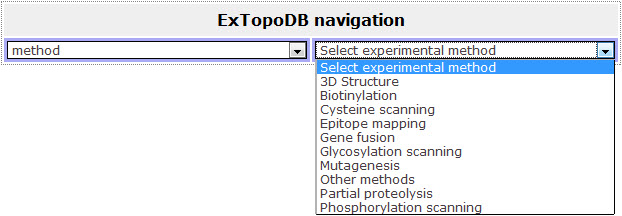
Figure 3. View of the drop down menu appearing when the user chooses to browse the database by Experimental methods.
By clicking on one of the available experimental methods the user is presented with an additional drop-down menu where he can now choose for the proteins of a specific organism for which this experimental method was used in order to determine their topologies.
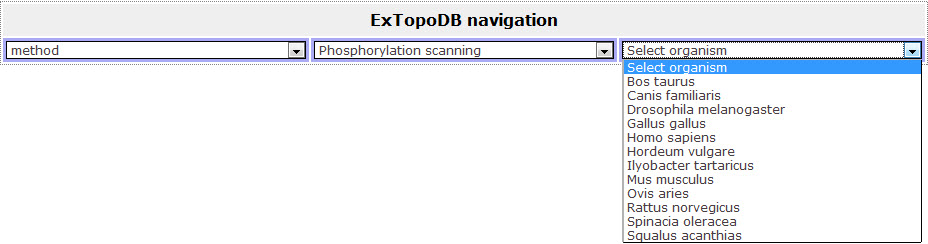
Figure 4. View of the drop down menu appearing when the user chooses one of the experimental methods. Here is the list of organisms available in the database whoose proteins' topology waw studied through Phosphorylation scanning.
Then by choosing one of the organisms he can get a list of the protein entries of ExtopoDB satisfying these criteria.
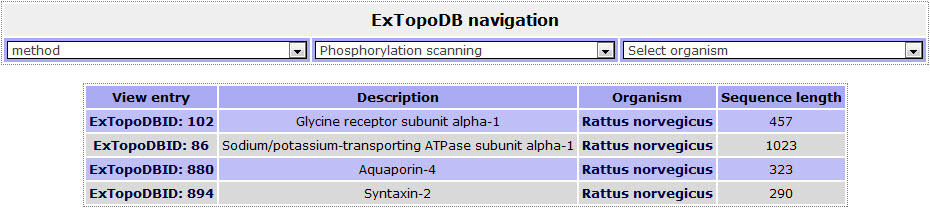
Figure 5. The list of proteins from the specific organism the user chose (Rattus norvegicus) whose topology was studied by the Phosphorylation scanning method.
If the user chooses to browse by Organisms he can choose from a drop-down list of organism names present in ExTopoDB.
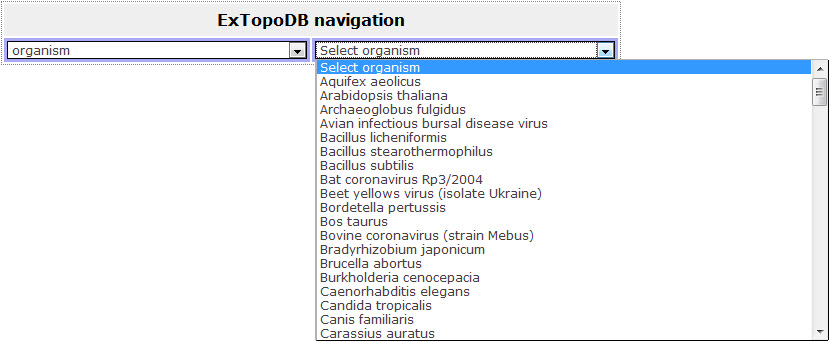
Figure 6. View of the drop down menu appearing when the user chooses to browse the database by Organisms.
By clicking on one of the available Organism names the user is presented with an additional drop-down menu where he can now choose for a specific experimetal method that was used for the determination of the topology of the proteins belonging to this organism and are present in ExTopoDB. Note that the additional drop-down list of Experimental methods includes only these methods that were used and not all.
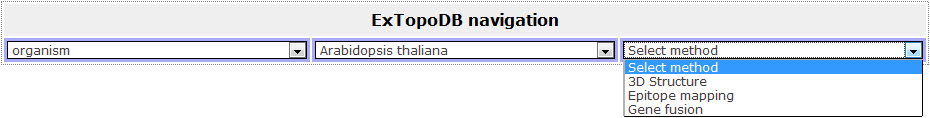
Figure 7. View of the drop down menu appearing when the user chooses one of the Organism names. Here is the list of experimental methods that were used for the determination of the topology of proteins of Arabidopsis thaliana. Notice that in this example only two methods (epitope mapping and gene fusion) were used for proteins of this organism that are included in ExTopoDB.
Then by choosing one of the Experimental methods he can get a list of the protein entries of ExtopoDB satisfying these criteria.
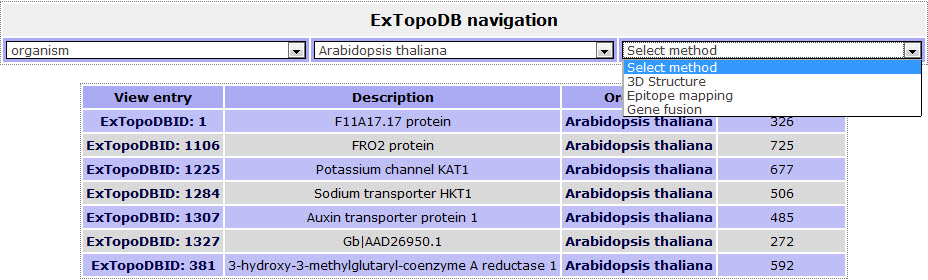
Figure 8. The list of proteins whose topology was studied by the specific experimental method the user chose (Gene fusion) and belong to Arabidopsis thaliana.
Return to top
Text Search
In the Text Search page, the user can search for any text in the fields of his/her preference. The user can enter any word (or expression) in one or more of the available boxes under the name: 'Protein Name', 'Gene name', ’Organism name’, 'Organism taxonomy', 'Cross - references' and 'Literature references'.
In the 'Experimental method' field the user can choose from several biochemical methods that were used for the determination of the protein's topology across the membrane.
Moreover, the user can submit a query that combines search expressions using the 'AND' operator or get results that satisfy at least one of the sub-expressions by checking the 'OR' operator. The user has also the ability to select if he wants to exclude fragments from the results.
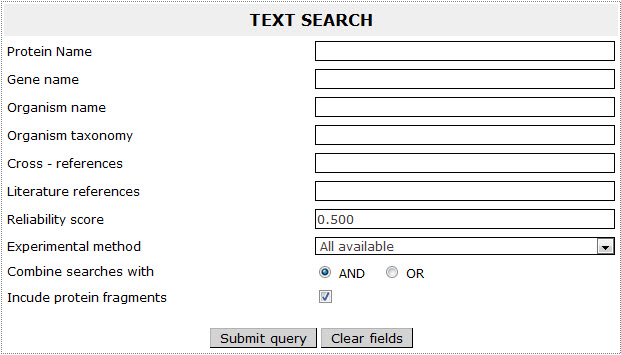
Figure 9. Basic view of the Text Search page.
Protein name
Corresponds to the field Protein description of an entry.
Example 1:
If the user wants to retrieve all Aquaporins included in ExTopoDB, has to use the name “aquaporin” as a query in the Protein name field:
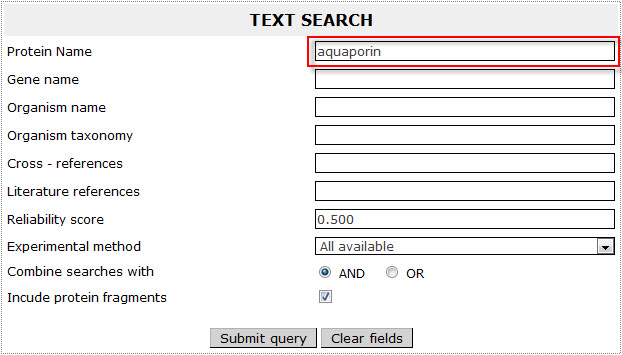
At the result's page the user can see a short description (ExTopoDB_ID, Uniprot Accession Number (AC), Protein name, Organism name and Sequence length ) of the entries matching the search criteria.
The result page for the above search is:
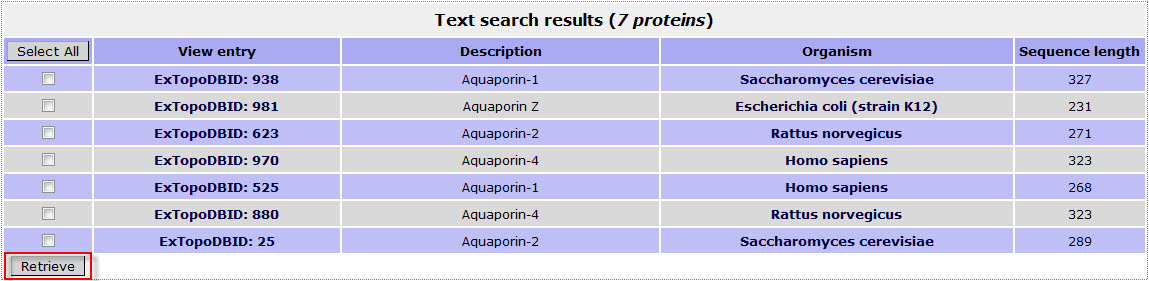
! Notice that the user can click the box of the Organism name and retrieve this way all proteins in the database from this specific organism.
! Notice that the user can check the box left to each entry and retrieve this way the entries that match the query by clicking the button "Retrieve".Then the user is directed to a page where he can choose from different file formats to download the selected entries.
Return to top
Gene name
Corresponds to the field Gene name of an entry.
Example 2:
If the user wants to retrieve all ATP synthase C chains included in ExTopoDB, has to use the name “atpE” as a query in the Gene name field:
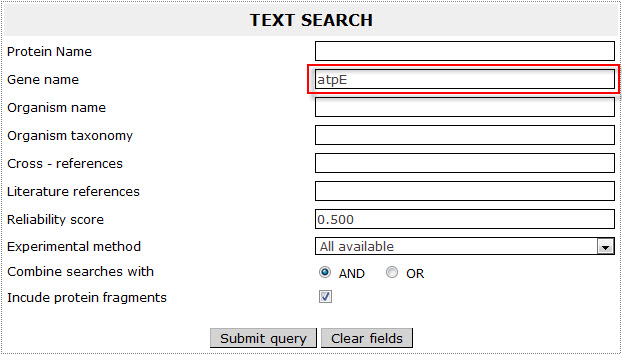
The result page for the above search is:
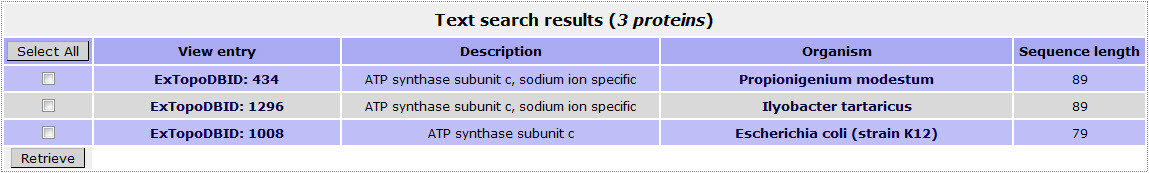
Return to top
Organism name
Corresponds to the field Species of an entry.
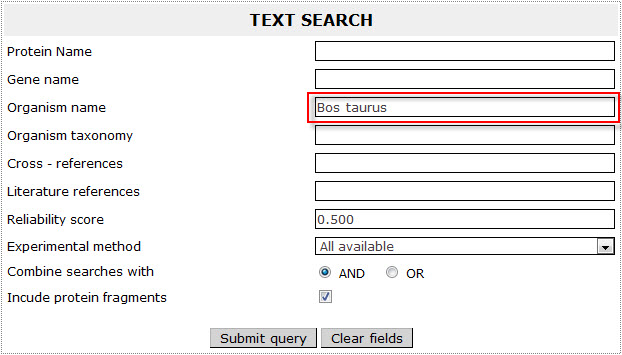
Example 3:
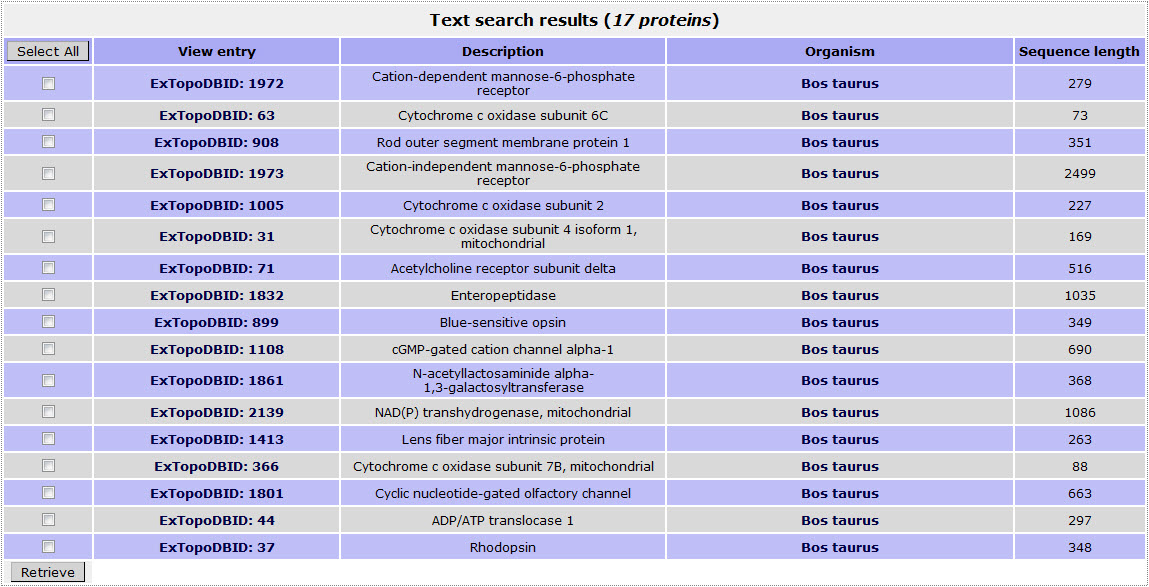
If the user wants to retrieve all bovine (Bos taurus) transmembrane proteins included in ExTopoDB, has to use the name “Bos taurus ” as a query in the Organism name field:
The result page for the above search is:
Return to top
Organism taxonomy
Corresponds to the field NCBI taxonomy of an entry.
Example 4:
If the user wants to retrieve all bovine (Bos taurus) transmembrane proteins included in ExTopoDB and knowns the NCBI taxonomy, has to use the name “9913 ” as a query in the Organism taxonomy field:
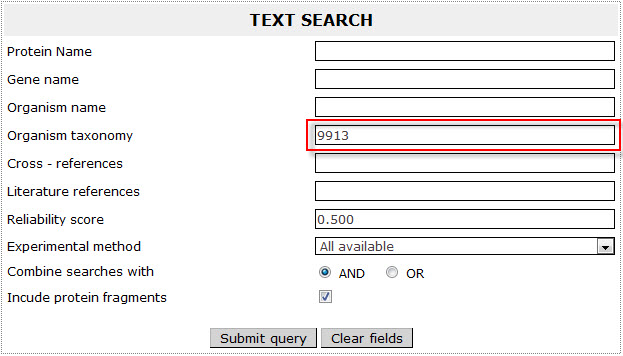
The result page for the above search is:
! Notice that the results are the same in the two examples when either using the Organism name (Example 3 ) or the NCBI taxonomy (Example 4).
Return to top
Cross - references
Corresponds to the field Cross - references field of an entry and in specific to the accession numbers or IDs of other selected publicly accessible biological databases.
Example 5:
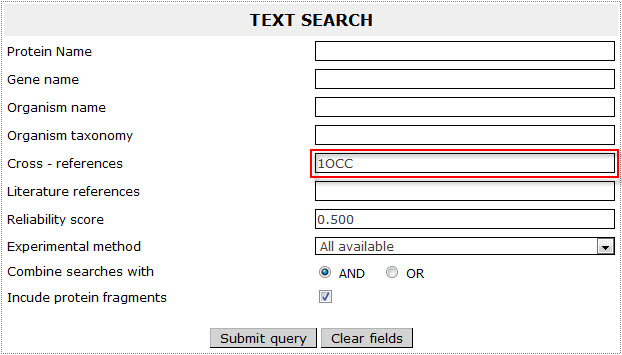
If the user wants to retrieve a protein included in ExTopoDB and has the accession number or ID of the protein in an other database (Uniprot, PFAM, InterPro, ProDom, PRINTS, TopDB, TCDB, PDB, EMBL, SMART, PROSITE, PIR, Psortdb), has to use this specific ID as a query in the Cross - references field.
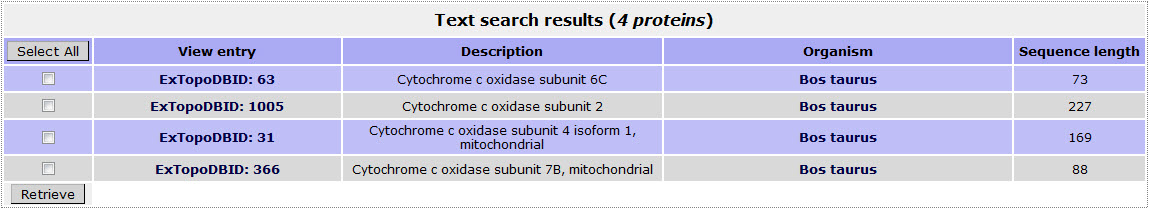
For example in order to retrieve proteins included in ExtopoDB that have the PDB code "1OCC" (STRUCTURE OF BOVINE HEART CYTOCHROME C OXIDASE AT THE FULLY OXIDIZED STATE ), the user has to use the name "1OCC" as a query in the Cross - references field:
The result page for the above search is:
Return to top
Literature references
Corresponds to the field Literature references of an entry and in specific to the Pubmed ID (PMID) of the literature reference(s) reporting the topological information included in each entry.
Example 6:
If the user wants to retrieve all proteins reported in a study with a specific PMID included in ExTopoDB, has to use the PMID as a query in the Literature references field:
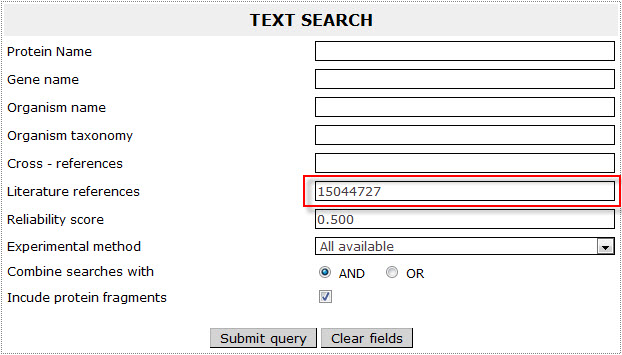
PMID corresponds to the study:
Rapp M, Drew D, Daley DO, Nilsson J, Carvalho T, Melιn K, De Gier JW, Von Heijne G.
Experimentally based topology models for E. coli inner membrane proteins.
Protein Sci. 2004 Apr ;13(4):937-45
The result page for the above search is:
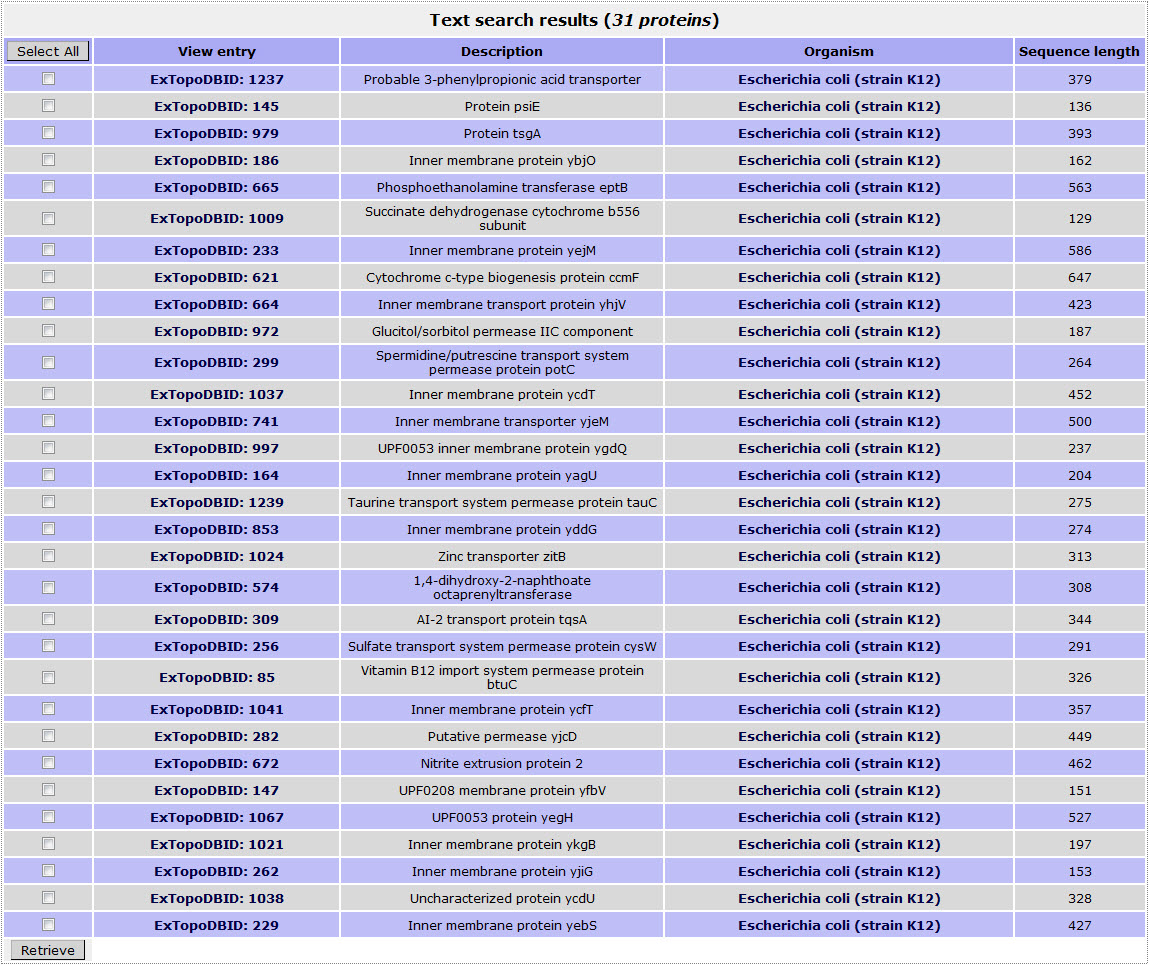
Return to top
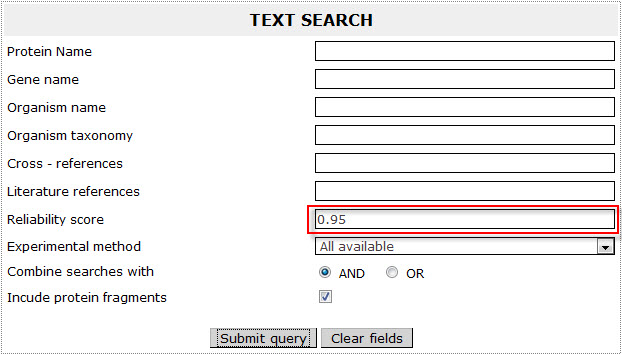
Reliability Score
Corresponds to the Reliability score of HMMTM's constrained prediction results. For instance the user can browse entries in ExTopoDB that have proposed topology through the HMMTM's constrained prediction results with a Reliablity score over 90% by giving the number 0.90 in the field.
Example 7:
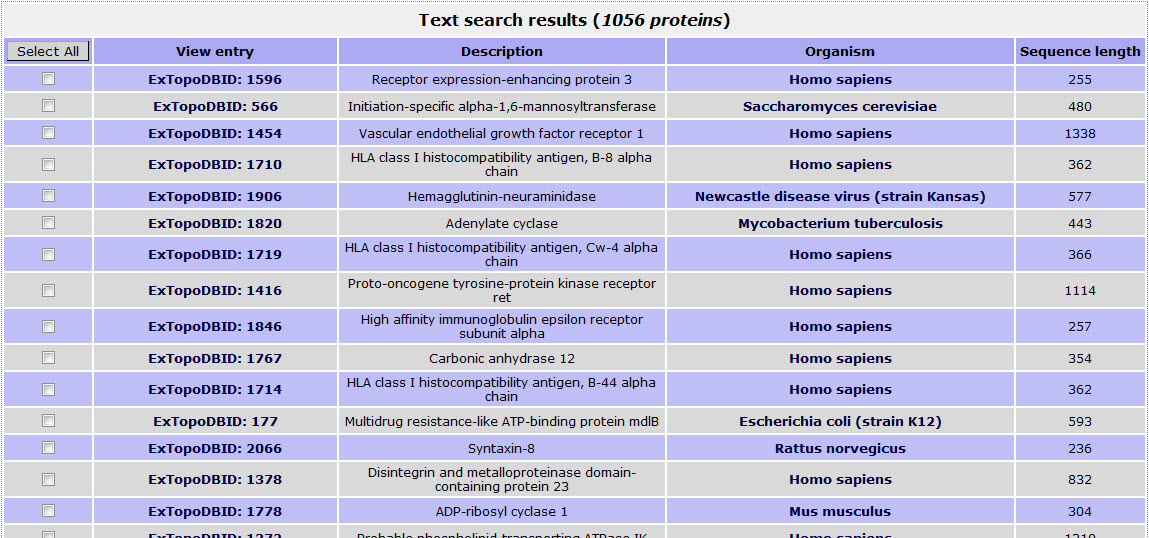
If the user wants to retrieve all proteins reported having proposed topology with a reliability score over 95%, has to give the number 0.95 in the field:
The top of the result page for the above search is:
Return to top
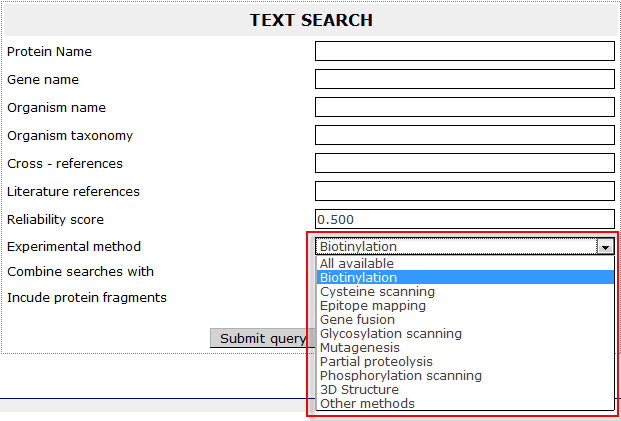
Experimental method
Corresponds to the Experimental method box of the Literature references field of an entry and the user can choose from pre-defined biochemical methods in a drop-down menu.
Example 8:
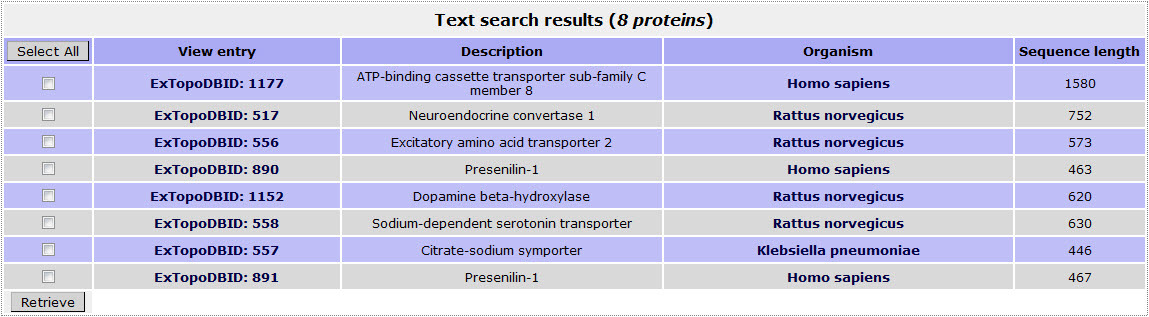
If the user wants to retrieve all proteins in ExTopoDB for which the topology determination was performed using the Biotinylation method, has to check the Biotinylation option the Experimental method field:
The result page for the above search is:
Return to top
BLAST search
With the BLAST search tool, the user may submit a sequence and search the database for finding homologues. The input for the BLAST application is the sequence in standard FASTA format and the user has the ability to specify an e-value cutoff level to use in the query:
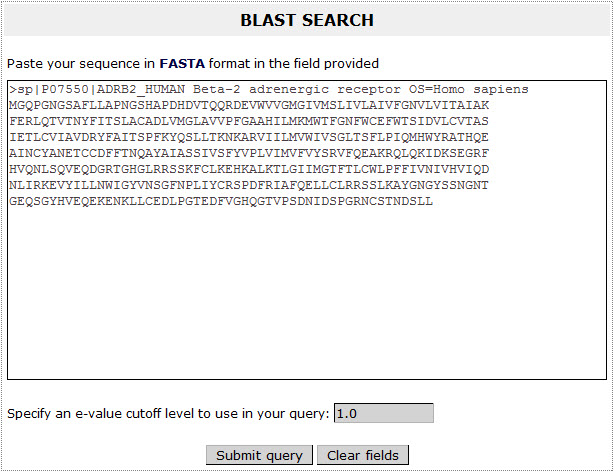
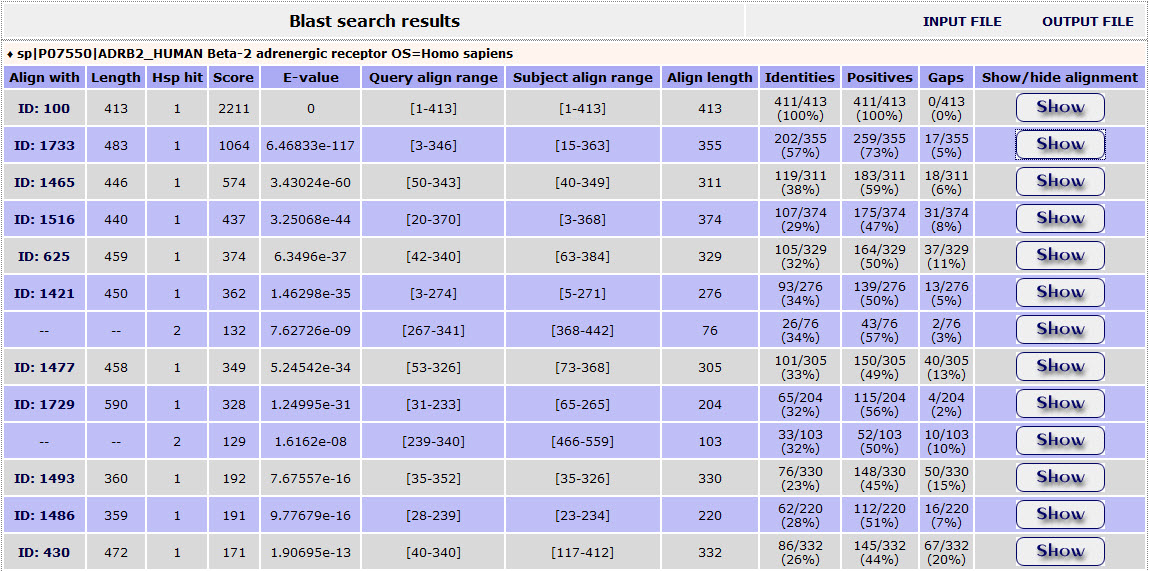
The result page of the BLAST search shows a list of the Blast hits with significant alignment on the query sequence the user has submited. The list is in a table format including the ExTopoDB_ID of the target protein, the Length of the target sequence and the Query and Target align range. The BLAST results can be compared through the Score and E-value and the Identities and Positives.
The result page of the above BLAST search is:
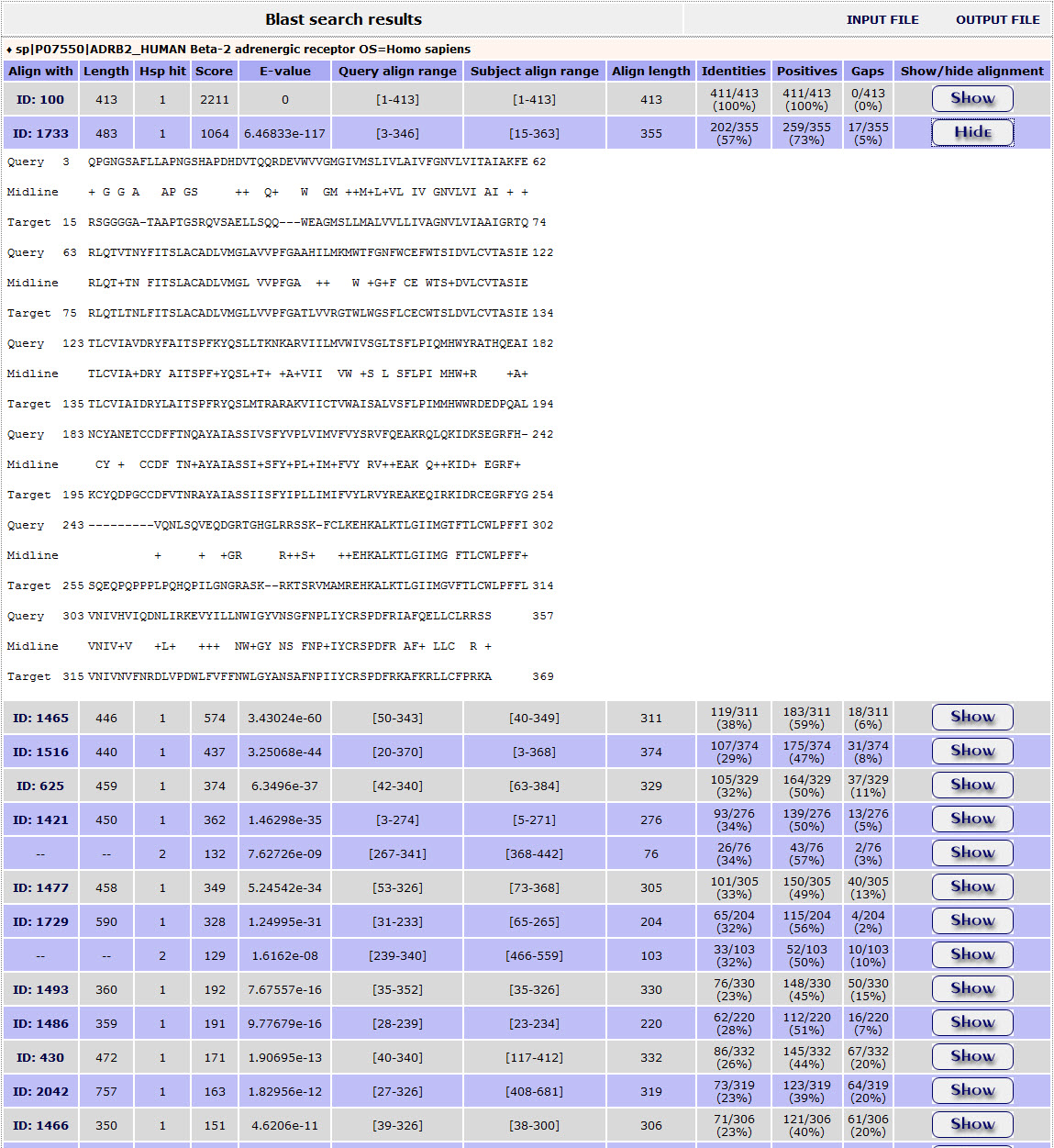
Furthermore, the user can have a more detailed view of each alignment through the Show/Hide button at the end of each line:
Return to top
Detailed view of an entry
Each entry in ExTopoDB has an unique ExTopoDB_ID number which is presented at the top of each entry (e.g. ::ExTopoDB 983::). Furthermore, the user can chose from other views of the entry besides of the basic view. These are: FASTA, TEXT, TOPOLOGY and XML. More details about these formats are described in the Download section of this manual.
The available fields in a protein entry of ExTopoDB are organized into six sections:
Firstly, the 'Protein details' section contains fields describing genelar information about the protein which are collected from Uniprot:
-
Protein description
-
Gene name
-
Species
-
Common name
-
NCBI taxonomy
-
Subcellulal localization
-
Sequence
-
Sequence length
-
Fragment: this field informs whether the protein is a fragment of not (Y/N).
Through the 'All proteins from this organism' button next to the 'Common name' field the user can get a list of all proteins in ExTopoDB that belong to this organism.
The 'Cross references' section contains fields corresponding to accesion number that link to other public available databases that contain information for the protein such as:
-
Uniprot
-
PFAM
-
InterPro
-
ProDom
-
PRINTS
-
TopDB
-
TCDB
-
PDB
-
EMBL
-
SMART
-
PROSITE
-
PIR
-
Psortdb
The 'Signal Peptide Information' section contains fields that give information about the existence of a signal peptide:
-
Amino acids: this field gives the begining and the end of the signal peptide in numbers relative to the length of the sequence.
-
Sequence: this field shows the sequence of the signal peptide only.
-
Source: this field informs the user whether the information for the existence of a signal peptide is from prediction methods (SignalP) of is experimentally verified.
The 'HMM-TM results' section contains the results of the constrained and unconstrained topology predictions generated with HMMTM. In both cases we provide the topology results along with a posterior probability plot showing the predicted transmembrane segments (red bars). For the constrained prediction we have incorporated for each protein the experimental information which we provide through ExTopoDB. Moreover, the user can customize the constrained prediction bye using experimental information of his interest through the 'User-customised HMM-TM prediction' field. All prediction results can be compared easily through the posterior probability plots and the Reliability score we provide.
The 'Topology information' section contains experimental information about the topology of each protein which was collected from the literature. Information is organized by reference and contains the number of the redidues and their localization relative to the membrane. Possible localizations of a residue can be: inside (I) , outside (O) , membrane (M) and part of a reentrant region (X).
The 'Literature references' section contains information about the literature references that were used for the 'Topology information' section. Literature references are organized in an ordered list where the user can see the author of the study, it's title, the journal and issue the study was published and at last the Pubmed ID (PMID) which links directly to Pubmed. Moreover, in the 'Experimental method' field we report the experimental method that was used in each study to generate the topological information we collected. Through the button 'All proteins from this reference' the user is presented with the list of proteins whose topology was studied in the particular refence. This is due to the fact that some studies described the use of biochemical methods for the determination of the topology of more than one different proteins.
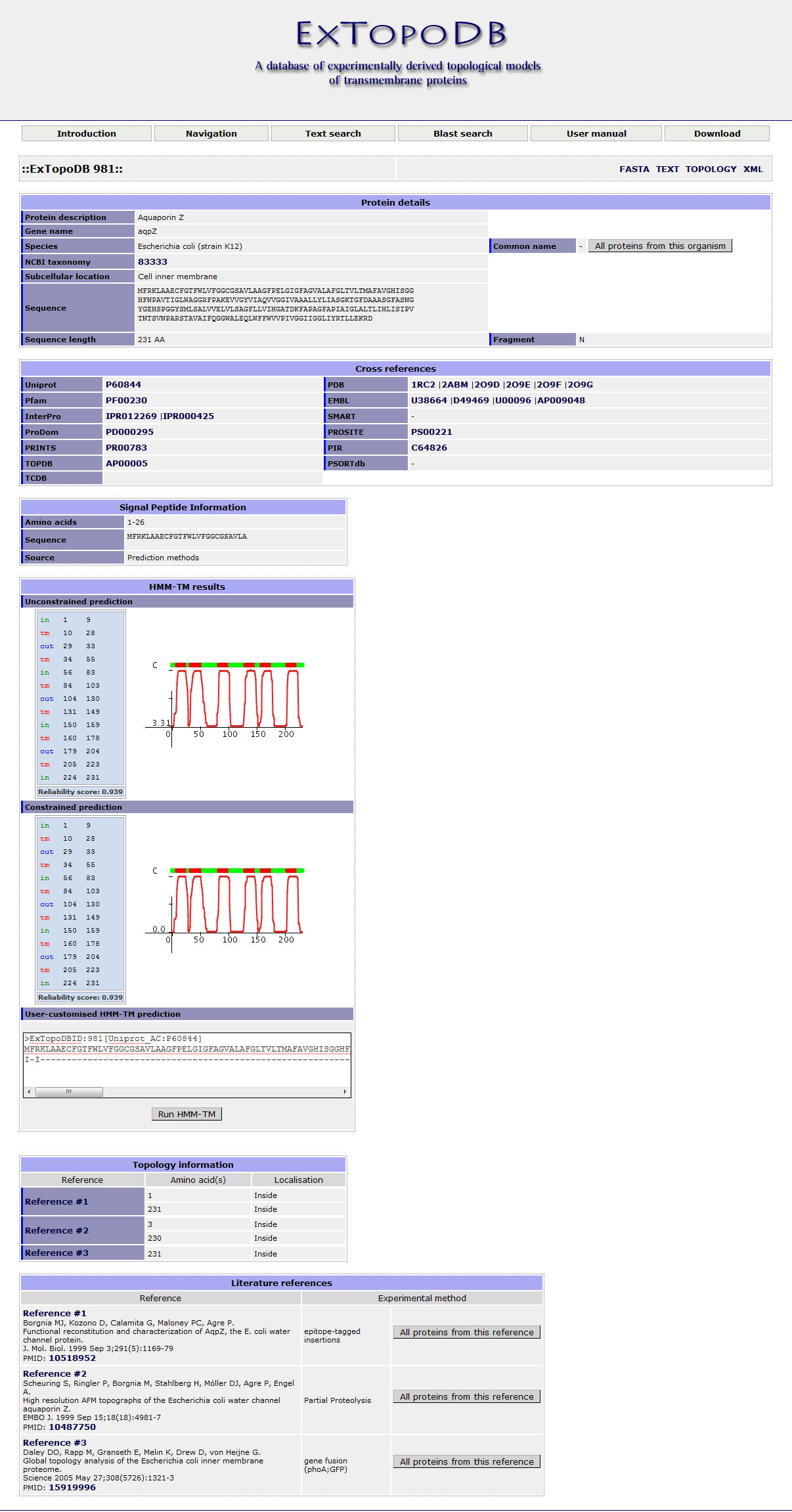
Figure 10. Detailed view of an ExTopoDB entry.

Figure 11. Detailed view of the same ExTopoDB entry in FASTA format.
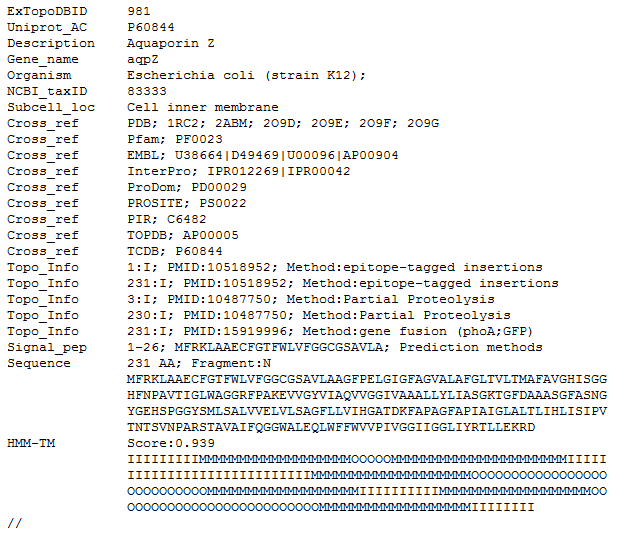
Figure 12. Detailed view of the same ExTopoDB entry in TEXT format.
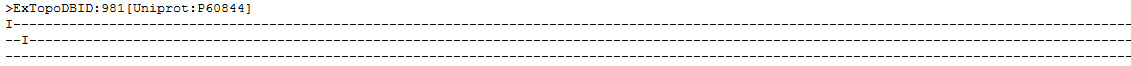
Figure 13. Detailed view of the same ExTopoDB entry in TOPOLOGY format.
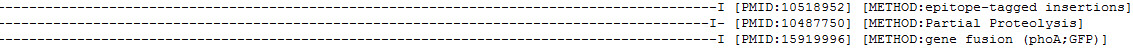
Figure 14. Detailed view of the same ExTopoDB entry in TOPOLOGY format (the end of the previous figure lines).
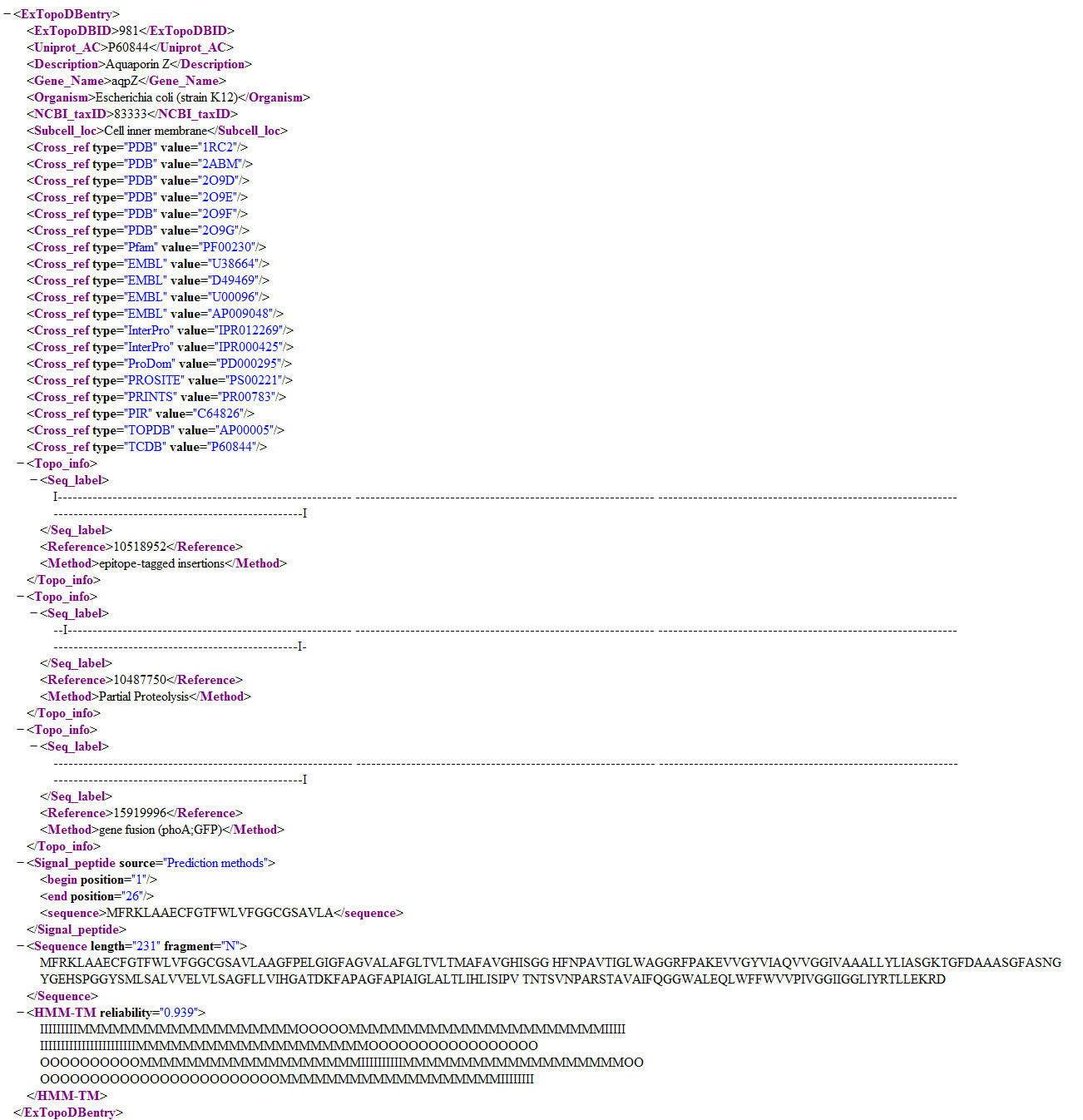
Figure 15. Detailed view of the same ExTopoDB entry in XML format.
Return to top
Download
Through the Download page the user can download the database in one of the following formats:

The ExTopoDB_v1 FASTA-format corresponds to the sequences of all proteins in ExTopoDB in FASTA format. In each sequence the ExTopoDB_ID and the Uniprot_AC are included:

The ExTopoDB_v1 FASTA-format [topology information included] corresponds to the sequences of all proteins in ExTopoDB in FASTA format followed by a line, corresponding to the topology information collected from the literature. Again, in each sequence the ExTopoDB_ID and the Uniprot_AC are included:

! Notice that after the line with the topology information of the protein, follows the PMID of the study of which this information was retrieved and the experimental METHOD that was used.
If in a protein entry there are more than one literature references providing information about the protein's topology then each literature reference corresponds to a different line and again the user can see the source of this information in the literature (PMID) and the type of the biochemical method for the determination of this topolgical information:

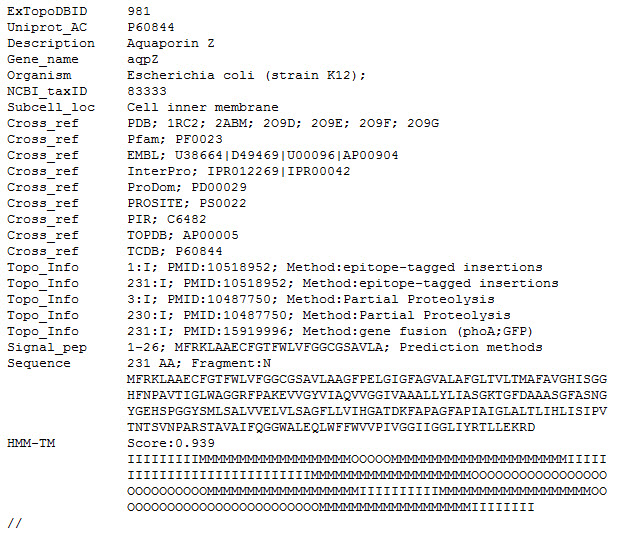
The ExTopoDB_v1 TEXT-format corresponds to all entries in ExTopoDB in simple text format. Entries are separated by "//" and all information contained in a typical ExtopoDB entry is encoded in simple text format allowing easier and faster data manipulation:
The ExTopoDB_v1 XML-format corresponds to all entries in ExTopoDB in Extensible Markup Language (XML) format:
Return to top