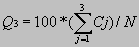 |
ΑΣΚΗΣΗ:
ΜΕΡΟΣ Α:
ΒΑΣΕΙΣ ΔΕΔΟΜΕΝΩΝ ΔΟΜΙΚΗΣ ΚΑΙ ΜΟΡΙΑΚΗΣ ΒΙΟΛΟΓΙΑΣ ΚΑΙ ΕΡΓΑΛΕΙΑ ΑΝΑΛΥΣΗΣ ΣΤΟ ΔΙΑΔΙΚΤΥΟ (INTERNET)
ΜΕΡΟΣ B:
ΠΡΟΓΝΩΣΗ ΔΟΜΗΣ ΔΙΑΜΕΜΒΡΑΝΙΚΩΝ ΠΡΩΤΕΪΝΩΝ
Β. Ι. Προμπονάς, Γ. Α. Παλαιός, Θ. Δ. Λιακόπουλος,
Ι. Σ. Χαμόδρακας, C. Pasquier και
Σ. Ι. Χαμόδρακας
Τμήμα Βιολογίας,
Τομέας Βιολογίας Κυττάρου και Βιοφυσικής,
Πανεπιστήμιο Αθηνών
Αθήνα, Απρίλιος 1999
Αναμφισβήτητα, οι πρωτεΐνες είναι από τα σπουδαιότερα βιολογικά μακρομόρια, με τεράστια ποικιλία βιολογικών δράσεων. Ενδεικτικά, υπάρχουν πρωτεΐνες που έχουν σημαντική ενζυμική δράση με μεγάλη εξειδίκευση (π.χ. ενδονουκλεάσες), άλλες είναι υπεύθυνες για τη δημιουργία διαφόρων τύπων υποδοχέων για νευροδιαβιβαστές, ορμόνες και άλλες ουσίες (π.χ. υποδοχέας ακετυλοχολίνης). Σε άλλες περιπτώσεις, σχηματίζουν μεγάλη ποικιλία ιοντικών καναλιών (π.χ. αντλία Κ+-Να+), αναλαμβάνουν τη μεταφορά - αποθήκευση ουσιών ζωτικής σημασίας (π.χ. αιμοσφαιρίνη, φερριτίνη) ή αποτελούν βασικά δομικά στοιχεία των κυττάρων (π.χ. κερατίνες).
Τα κάθε μορφής δεδομένα σχετικά με πρωτεΐνες που αποτελούν προϊόν πειραματικής έρευνας ή υπολογιστικής ανάλυσης, κατατίθενται σε βάσεις δεδομένων, αρκετές από τις οποίες διατίθενται για ελεύθερη πρόσβαση στην Ακαδημαϊκή Κοινότητα. Η ευρύτατη χρήση του διαδικτύου (INTERNET) τα τελευταία χρόνια, έχει καταστήσει πολύ απλή και γρήγορη την πρόσβαση των ερευνητών σε μεγάλη ποικιλία πληροφοριών. Ταυτόχρονα προσφέρεται πρόσβαση σε μεγάλο αριθμό προγραμμάτων λογισμικού, που πραγματοποιούν διάφορα είδη υπολογιστικής ανάλυσης.
Στο URL:
http://srs.ebi.ac.uk/srs6bin/cgi-bin/wgetz?-page+top+-newId
βρίσκεται ένας κατάλογος που περιέχει βασικές πληροφορίες για μια αρκετά ευρεία συλλογή βάσεων δεδομένων που συσχετίζονται με πρωτεΐνες. Επιλέγοντας τις αντίστοιχες συνδέσεις (links) έχουμε πρόσβαση σε λεπτομερέστερες πληροφορίες για κάθε μία από αυτές.
Οι παραπάνω βάσεις δεδομένων είναι δυνατόν να περιέχουν απλά συλλογές πρωτεϊνικών ακολουθιών ή ακολουθίες συνοδευόμενες από συμπληρωματικά σχόλια. Στην τελευταία κατηγορία ανήκει η SWISS-PROT (Bairoch and Apweiler, 1998), με 77977 καταχωρήσεις (στις αρχές του 1999) στις οποίες μπορούν, επιπλέον, να βρεθούν βιβλιογραφικές αναφορές, γενικά στοιχεία δευτεροταγούς δομής καθώς και σημειώσεις για τη βιολογική λειτουργία (αν είναι γνωστές) και άλλες χρήσιμες πληροφορίες.
Η Protein Data Bank (PDB, Sussman et al., 1998) αποτελεί μία αρχειοθέτηση πειραματικά προσδιορισμένων Τρισδιάστατων Δομών Βιολογικών Μακρομορίων. Στα αρχεία της βρίσκονται ταξινομημένες πληροφορίες σχετικές με τις δομές, όπως ατομικές συντεταγμένες, βιβλιογραφικές αναφορές, στοιχεία της Πρωτοταγούς και Δευτεροταγούς δομής, καθώς και λεπτομερή δεδομένα κρυσταλλογραφίας ακτίνων-Χ ή δεδομένα φασματοσκοπίας NMR. Σήμερα, 9809 δομές πρωτεϊνών και νουκλεϊκών οξέων βρίσκονταί καταχωρημένες στην PDB. Αν αυτός ο αριθμός συγκριθεί με τις γνωστές ακολουθίες πρωτεϊνών, γίνεται εμφανές ότι δημιουργείται ένα 'χάσμα' ανάμεσα στις πειραματικά προσδιορισμένες δομές και τις γνωστές αμινοξικές ακολουθίες (Rost. and Sander, 1996).
Περισσότερο εξειδικευμένες πληροφορίες, εξαιρετικά σημαντικές για την πιο μεθοδική μελέτη των πρωτεϊνών, ταξινομούνται σε άλλες βάσεις δεδομένων. Για παράδειγμα η CATH (Orengo et al., 1997) περιέχει μια ιεραρχική ταξινόμηση Αυτοτελών Δομικών Στοιχείων (Domains), πρωτεϊνικών δομών κατατεθειμένων στην PDB, οι οποίες έχουν προσδιοριστεί σε διακριτικότητα καλύτερη από 3 A. Μη πρωτεϊνικές δομές, μοντέλα και Cα δομές δεν έχουν συμπεριληφθεί. Τέσσερα κύρια επίπεδα χρησιμοποιούνται στην ταξινόμηση αυτή: τα επίπεδα Τάξης (Class), Αρχιτεκτονικής (Architecture), Τοπολογίας (fold family) και Ομόλογης Υπέρ-οικογένειας (Homologous superfamily).
Επίσης, μετά από μελέτες ομολογίας πρωτεϊνών της ίδιας λειτουργίας έχουν εντοπιστεί διατηρημένα μοτίβα στις αμινοξικές τους ακολουθίες (Patterns), τα οποία χαρακτηρίζουν τις πρωτεΐνες της οικογένειας και είναι ταξινομημένα στη βάση δεδομένων PROSITE (Bairoch et al, 1997).
Είναι πλέον γνωστό ότι η λειτουργικότητα (βιολογική δράση) των πρωτεϊνών εξαρτάται κυρίως από την στερεοδιάταξη τους (δομή τους στο χώρο), από τον τρόπο δηλ. με τον οποίο η γραμμική αμινοξική τους ακολουθία αναδιπλώνεται στο χώρο. Η τρισδιάστατη δομή μερικών χιλιάδων πρωτεϊνών σε ατομική ή περίπου ατομική διακριτικότητα, αποκαλύφθηκε με μεθόδους κρυσταλλογραφίας ακτίνων-Χ μονοκρυστάλλων (Sussman et al., 1998). Αυτές οι μέθοδοι είναι δαπανηρές, επίπονες και χρονοβόρες. Επίσης απαιτούν τη χρήση μονοκρυστάλλων, που δεν δημιουργούνται εύκολα για αρκετές πρωτεΐνες. Επομένως, προέκυψε η ανάγκη να ληφθούν πληροφορίες για το πρωτεϊνικό δίπλωμα με άλλες μεθόδους.
Τις τελευταίες δύο δεκαετίες, οι αμινοξικές ακολουθίες περισσότερων από 70.000 πρωτεϊνών είναι γνωστές (Bairoch and Apweiler, 1998). Αφού τα πειραματικά δεδομένα σαφέστατα δείχνουν ότι όλη η αναγκαία πληροφορία ώστε μια πρωτεΐνη να διπλωθεί στην φυσική της στερεοδομή είναι κωδικοποιημένη στην γραμμική αμινοξική της ακολουθία (Anfinsen, 1973), έγιναν πολλές προσπάθειες ώστε να προβλεφθεί η δομή μια πρωτεΐνης από την αμινοξική της ακολουθία (Rost and Sander, 1994; Rost and Sander, 1996), αλλά με περιορισμένη επιτυχία μόνο. Επίσης, πρόσφατα, τα λεγόμενα προγράμματα προσδιορισμού της πλήρους ακολουθίας του DNA (γονιδιώματος) για πολλούς οργανισμούς, μεταξύ των οποίων και του ανθρώπου (genome projects), επομένως και όλων των πρωτεϊνών που κωδικοποιεί, έχουν ως αποτέλεσμα να συσσωρεύονται τεράστια ποσά πληροφοριών σχετικά με τις ακολουθίες χιλιάδων πρωτεϊνών , με ιλιγγιώδεις ρυθμούς (Bork et al. 1994). Ένα μεγάλο ποσοστό (40-50%) αυτών των πρωτεϊνών που προσδιορίζονται από την αναγνώριση των ORF (Open Reading Frames, γονιδίων) είναι τελείως άγνωστης λειτουργίας. Η Τεχνολογική Επανάσταση στον τομέα των Υπολογιστικών Συστημάτων και της Πληροφορικής, παρέχει σήμερα αξιόλογα εφόδια για τη δημιουργία νέων αλλά και τη βελτίωση παλαιών προγνωστικών αλγορίθμων και μεθόδων.
Γενική πρόγνωση της δομής από την ακολουθία δεν έχει επιτευχθεί ακόμη, αφού δεν έχει καταστεί δυνατή η απόλυτη συσχέτιση ακολουθίας-δομής. Παρά το γεγονός αυτό, αποκαλύπτονται διαρκώς κάποιοι κανόνες, αξιοποιώντας τις ολοένα αυξανόμενες πληροφορίες από πειραματικά προσδιορισμένες πρωτεϊνικές δομές.
Αρχικά, γίνεται εμφανές ότι ο αριθμός των διακεκριμένων προτύπων για το δίπλωμα είναι πεπερασμένος (Chothia C., 1992; Finkelstein A.V.and Reva B.A, 1992): μάλιστα είναι εξαιρετικά μικρότερος από αυτόν που θα ανέμενε κανείς, λόγω της αφθονίας των διαφορετικών συνδυασμών με τους οποίους μπορούν αμινοξικά κατάλοιπα να συνθέσουν πολυπεπτιδικές αλυσίδες (π.χ. για αλληλουχία 100 καταλοίπων υπάρχουν 20
Βασισμένοι σε αυτό τον υπολογισμό και στις πρόσφατες αναλύσεις ολόκληρων χρωμοσωμάτων - γονιδιωμάτων, εκτιμάται ότι ο συνολικός αριθμός διαφορετικών διπλωμάτων είναι της τάξης του 103.
Αφού τα πειραματικά δεδομένα οδηγούν με όλο και μεγαλύτερη βεβαιότητα στη διαπίστωση ότι η απαραίτητη πληροφορία για το δίπλωμα μιας πρωτεΐνης υπάρχει στην πρωτοταγή της δομή (Anfinsen, 1973), πολλές προσπάθειες έχουν γίνει μέχρι σήμερα για την πρόγνωση της 3D-δομής (και κατ΄ επέκταση της λειτουργικότητας) των πρωτεϊνών από την ακολουθία και μόνο. Σημαντικό γεγονός, που ενισχύει τη θεώρηση αυτή, είναι η παρατήρηση πως πρωτεΐνες με πολύ μικρή ομολογία (μικρότερη από 30%) σε επίπεδο αμινοξικής ακολουθίας, φαίνεται να έχουν το ίδιο δίπλωμα στο χώρο και την ίδια λειτουργία. Στην πράξη, η μεγάλη πλειοψηφία ζευγών πρωτεϊνών με παρόμοιες δομές παρουσιάζουν σχετικά χαμηλή ομολογία στην πρωτοταγή τους δομή (Rost, 1997, Rost, et al., 1998). Δηλαδή, η πρωτεϊνική δομή είναι περισσότερο διατηρημένη από την ακολουθία (Chothia and Lesk, 1986, Doolittle, 1986, Lesk, 1991).
Παρ΄ όλα αυτά, είναι δυνατόν πρωτεΐνες όμοιες σε μεγάλο βαθμό να έχουν εντελώς διαφορετικούς βιολογικούς ρόλους. Χαρακτηριστικό παράδειγμα είναι η κρυσταλλίνη στο φακό του οφθαλμού (με καθαρά δομικό ρόλο) η οποία είναι εμφανές προϊόν πρόσφατης εξέλιξης από μεταβολικά ένζυμα (Piatigorsky and Wistow, 1989). Επίσης, σε αρκετές περιπτώσεις, δομική ή ακόμη και λειτουργική ομοιότητα εμφανίζεται για περιορισμένα τμήματα (domains : αυτοτελή δομικά στοιχεία) πρωτεϊνών. Για παράδειγμα η ύπαρξη προτύπου "δακτύλου ψευδαργύρου" (Zink-finger motif), είναι μια καλή ένδειξη πως η συγκεκριμένη περιοχή είναι δυνατό να προσδένεται σε DNA ή RNA, δε μπορούμε, όμως, να αποκλείσουμε απόλυτα την πιθανότητα ώστε, συνολικά, η πρωτεΐνη να συμμετέχει σε κάποια άλλη βιολογική λειτουργία. Συνεπώς, οφείλει κανείς να είναι ιδιαίτερα προσεκτικός όταν προσπαθεί να επεκτείνει τα συμπεράσματά του και στη λειτουργία των πρωτεϊνών.
Συνδυασμοί στοιχείων δευτεροταγούς δομής, με συγκεκριμένη γεωμετρική διευθέτηση, εμφανίζονται συχνά σε δομές πρωτεϊνών. Τέτοιοι συνδυασμοί αποτελούν την υπερ-δευτεροταγή δομή και ορισμένοι από αυτούς συσχετίζονται άμεσα με συγκεκριμένες λειτουργίες. Για παράδειγμα, ο συνδυασμός helix-loop-helix αποτελεί ένα μοτίβο για πρόσδεση σε DNA (Gibson et al., 1993). Έτσι, επιτυχής πρόγνωση των στοιχείων δευτεροταγούς δομής δύναται να βοηθήσει στην αποκάλυψη τέτοιων σχηματισμών ανώτερης τάξης που παραπέμπει, συχνά, σε καθορισμένους βιολογικούς ρόλους.
Τα τελευταία χρόνια, με τον προσδιορισμό αλληλουχιών από αρκετά γονιδιώματα, το 'χάσμα' μεταξύ των αμινοξικών ακολουθιών που βρίσκονται κατατεθειμένες σε ελεύθερα προσβάσιμες τράπεζες δεδομένων, π.χ. SWISS-PROT και των λυμένων κρυσταλλογραφικά πρωτεϊνικών δομών (κατατεθειμένων στην PDB) διαρκώς αυξάνεται. Στις αρχές του 1998, γνωρίζαμε ήδη όλες τις ακολουθίες για δώδεκα ολόκληρα γoνιδιώματα (Gaasterland, 1997), ενώ τη στιγμή που γραφόταν η άσκηση 21 πλήρη γονιδιώματα βρίσκονται κατατεθειμένα σε βάσεις δεδομένων με ελεύθερη πρόσβαση, (Kyrpides, 1999). Αναφέρεται, ότι στην προσπάθεια να γεφυρωθεί αυτό το χάσμα η πιο επιτυχής θεωρητική προσέγγιση είναι το 'Homology Modelling', ενώ πολύ ελπιδοφόρο για το μέλλον το 'Threading'. Στη συνέχεια, παρουσιάζονται οι βασικές αρχές αυτών των μεθόδων και τα αποτελέσματά τους, τα οποία αν και εντυπωσιάζουν με την πρώτη ματιά, πρέπει να μην υπερεκτιμούνται.
Εφαρμόζεται σε μια πρωτεΐνη άγνωστης δομής, αν βρεθεί ομόλογή της με λυμένη δομή.
Ανάλυση των στοιχίσεων μεταξύ ακολουθιών με γνωστές δομές αναδεικνύει ότι ζευγάρια πρωτεϊνών με μεγαλύτερη ομοιότητα από 30% (για μήκη στοίχισης > 80 καταλοίπων) (Eisenberg et al., 1984) έχουν ομόλογες 3D-δομές, δηλαδή το βασικό δίπλωμά τους είναι όμοιο και μόνο περιοχές με μη καθορισμένη δευτεροταγή δομή ίσως να διαφέρουν. Αν, λοιπόν, διαπιστωθεί ομολογία ανάμεσα σε μια πρωτεΐνη (U) άγνωστης 3D-δομής με μία γνωστής (Τ) τότε μπορεί να μοντελοποιηθεί μια δομή για την (U) με 'εκμαγείο' την (Τ).
Με τον τρόπο αυτό έγινε δυνατόν να μεγαλώσει το πλήθος των 'γνωστών' 3D-δομών από 7.000 σε περίπου 50.000 (Rost and Sander, 1996). Η βασική προϋπόθεση είναι ότι η (U) και η (Τ) έχουν ταυτόσημες κύριες αλυσίδες. Η δυσκολία βρίσκεται στη σωστή προσαρμογή των πλευρικών αλυσίδων, πρόβλημα του οποίου η επίλυση επιβάλει τη χρήση ενεργειακών υπολογισμών, καθώς και δεδομένων για όλες τις διευθετήσεις πλευρικών αλυσίδων σε λυμένες πρωτεϊνικές δομές.
Στην περίπτωση που δεν εμφανίζεται υψηλή ομολογία, τεχνικές 'Threading' (ή Remote Homology Modelling, όπως αλλιώς αναφέρονται) είναι δυνατόν να αποκαλύψουν 'απομακρυσμένες' ομολογίες, οι οποίες δεν ανιχνεύονται με τη χρήση συμβατικών μεθόδων στοίχισης ακολουθιών.
Με δεδομένη την ύπαρξη πολλών πρωτεϊνών στην PDB με ομολογίες μικρότερες από 25% αλλά 'ομόλογες' δομές, ορίστηκε η έννοια της 'απομακρυσμένης ομολογίας'. Η ιδέα είναι να ανιχνευθεί αυτή η απομακρυσμένη ομολογία και κατά τον ίδιο τρόπο με τη μέθοδο του Homology Modelling να μοντελοποιηθεί μια δομή για την άγνωστη πρωτεΐνη. Υπάρχουν διαφορετικά είδη τέτοιων μεθόδων, που ξεχωρίζουν μεταξύ τους κυρίως ως προς τον τρόπο προσδιορισμού της 'ομολογίας'.
Για να επιτύχει η διαδικασία του Threading είναι αναγκαίο να ξεπεραστούν 3 κύριες δυσκολίες:
1. Διαπίστωση της απομακρυσμένης ομολογίας
2. Ορθή στοίχιση των ακολουθιών
3. Χαμηλή πραγματική ομολογία, που δυσκολεύει την εύρεση της σωστής δομής, ιδίως στα τμήματα με μη κανονική δευτεροταγή δομή.
Απομακρυσμένες ομολογίες ανιχνεύονται πολύ συχνά, όμως καμία μέθοδος δεν κατορθώνει να εντοπίσει περισσότερες από τις μισές από αυτές τις περιπτώσεις σε μια προεπιλεγμένη ομάδα πρωτεϊνών που χρησιμοποιείται για αξιολόγηση των μεθόδων (Lemer, C. et al., 1995).
Παρόλο που οι μέθοδοι του 'Homology Modelling' και του 'Threading' δίνουν ορισμένες φορές εντυπωσιακά αποτελέσματα, δεν είναι δυνατό να θεωρήσουμε πως έχει λυθεί το πρόβλημα του διπλώματος των πρωτεϊνών. Ακόμη κι αν θεωρήσουμε πως υπάρχει μεγάλο ποσοστό επιτυχίας στην εύρεση των ομόλογων ακολουθιών, το παραμικρό σφάλμα κατά τη στοίχισή τους δε γίνεται να διορθωθεί στο τρίτο στάδιο της μοντελοποίησης. Αν και οι μέθοδοι αυτές αφήνουν μεγάλες υποσχέσεις για το μέλλον, είναι επικίνδυνο να υπερεκτιμηθούν τα αποτελέσματά τους. Για το λόγο αυτό, πολύ συχνά καταφεύγουμε σε απλουστευμένες προγνώσεις, που μπορεί να μη δίνουν 3D-δομές αλλά αποκαλύπτουν συχνά σημαντικά μυστικά για τη δομή των πρωτεϊνών.
Για τις περισσότερες πρωτεΐνες δεν είναι εφαρμόσιμες οι μέθοδοι του 'Homology Modelling' και του 'Threading' και η πρόγνωση που επιθυμούμε πρέπει να γίνει σε ένα απλοποιημένο επίπεδο: Μονοδιάστατες (1D) προγνώσεις (δευτεροταγούς δομής, διαμεμβρανικών τμημάτων, έκθεσης στο διαλύτη) είναι δυνατές, με αξιόλογα πολλές φορές αποτελέσματα. Σύμφωνα με πρόσφατες εργασίες, φαίνεται πιθανό ότι με τη χρήση 'εξελικτικής' πληροφορίας η απόδοσή τους βελτιώνεται αισθητά. Δισδιάστατες (2D) προγνώσεις (αλληλεπίδρασης μεταξύ απομακρυσμένων στην πρωτοταγή δομή καταλοίπων, δισουλφιδικών δεσμών) έχουν, προς το παρόν, μικρή ακρίβεια.
Η κεντρική ιδέα, γύρω από την οποία περιστρέφονται οι περισσότερες μέθοδοι πρόγνωσης δευτεροταγούς δομής, είναι το γεγονός ότι τμήματα διαδοχικών καταλοίπων έχουν προτιμήσεις να βρίσκονται σε συγκεκριμένες καταστάσεις δευτεροταγούς δομής (Szent-Gyοrgyi and Cohen, 1957, Blout et al., 1960, Scheraga, 1960, Chou and Fasman, 1978, Garnier et al., 1978). Επομένως, το πρόβλημα στην ουσία ανάγεται σε πρόβλημα αναγνώρισης προτύπων (pattern recognition) και αντιμετωπίζεται με τη χρήση αντίστοιχων αλγορίθμων. Μία συνηθισμένη προσέγγιση στο ζήτημα είναι να επιτευχθεί πρόγνωση για το κεντρικό κατάλοιπο μιας περιοχής με τυπικό εύρος 11-25 γειτονικών καταλοίπων. Συνήθως, η πρόγνωση γίνεται μεταξύ τριών καταστάσεων:
| 1. α-έλικα | (α-helix => H) |
| 2. β-πτυχωτή επιφάνεια | (β-sheet => Ε) |
| 3. τίποτε απ΄τα παραπάνω | ('coil' ή 'loop'=> L) |
Πολλοί διαφορετικοί αλγόριθμοι αναπτύχθηκαν γι' αυτό το σκοπό, βασιζόμενοι σε φυσικοχημικές αρχές, θεωρεία γράφων, στατιστική, νευρωνικά δίκτυα, πολλαπλές στοιχίσεις ακολουθιών κ.α., αλλά η επιτυχία τους έχει αποδειχθεί μάλλον περιορισμένη (Kabsch and Sander, 1983a). Στην πράξη έχει φανεί πως συνδυαστικές μέθοδοι πρόγνωσης δίνουν μεγαλύτερα ποσοστά επιτυχίας από τις μεμονωμένες μεθόδους (Schultz et al., 1974; Argos et al., 1976).
Οι μέθοδοι αξιολογούνται ως προς την αξιοπιστία τους με βάση λυμένες δομές πρωτεϊνών, κατατεθειμένων στην PDB. Ο προσδιορισμός της δευτεροταγούς δομής μπορεί να πραγματοποιηθεί για γνωστές 3D-δομές λαμβάνοντας υπ' όψη τη θέση στο χώρο των ατόμων που την απαρτίζουν, καθώς και το 'πρότυπο σχηματισμού Η-δεσμών' του καρβονυλίου (C=O) της κύριας αλυσίδας και των αμιδικών ομάδων (ΝΗ). Η μέθοδος DSSP (Kabsch and Sander, 1983b) καθορίζει 8 διαφορετικά στοιχεία δευτεροταγούς δομής τα οποία ανάγονται στις 3 γενικές κλάσσεις Η,Ε,L, που αναφέρθηκαν προηγούμενα, όπως φαίνεται στον ΠΙΝΑΚΑ 1,
με τις επόμενες παραδοχές:
Β- => ΕΕ , ενώ Β-Β => LLL
Είναι σημαντικό να παρατηρήσει κανείς ότι, αλλαγή στην αντιστοίχιση των κλάσεων της DSSP επιφέρει αλλαγή και στην εκτιμούμενη αξιοπιστία, η οποία μπορεί να υπολογιστεί:
Ανά-κατάλοιπο (τριών-καταστάσεων) : Eίναι ο απλούστερος και ευρύτερα διαδεδομένος τρόπος. Δίνεται από τη σχέση
|
όπου Cj το πλήθος των καταλοίπων που προβλέφθηκαν σωστά στην κατάσταση j(H,E,L) και Ν το συνολικό πλήθος καταλοίπων της ακολουθίας.
Ανά-στοιχείο δευτεροταγούς δομής : Γίνεται με
Α) Μέτρηση του συνολικού πλήθους στοιχείων στην πρωτεΐνη.
Β) Υπολογισμό του μέσου μήκους τους,
και
Γ) Εύρεση της κατανομής του πλήθους στοιχείων σε συνάρτηση με το μήκος (Rost and Sander, 1993).
Τα τρία αυτά αποτελέσματα συσχετίζονται, δίνοντας χρήσιμες επιπλέον πληροφορίες από την ανά-κατάλοιπο εκτιμούμενη αξιοπιστία και χαρακτηρίζουν τα αποτελέσματα των προγνώσεων, ιδιαίτερα σε περιπτώσεις προγνώσεων με υψηλό δείκτη Q3 που δεν αντιστοιχούν σε πραγματική κατανομή των προβλεφθέντων τμημάτων. Σήμερα, οι μέθοδοι πρόγνωσης δευτεροταγούς δομής δίνουν αποτελέσματα με Q3 < 70%, ποσοστό που αν και φαίνεται μικρό αποκαλύπτει συχνά πολύ σπουδαίες δομικές και, κατά συνέπεια, λειτουργικές πληροφορίες.
Μία πρώτη κατάταξη των πρωτεϊνών θα μπορούσε να διαχωρίσει τις πρωτεΐνες των οργανισμών σε σφαιρικές-υδατοδιαλυτές, σε δομικές, σε πρωτεΐνες με ιδιότητες ανάμεικτες με τις προηγούμενες δύο κατηγορίες (δομικές-σφαιρικές) και σε πρωτεΐνες μεμβρανών. Η τελευταία κατηγορία είναι εξαιρετικά ενδιαφέρουσα για πολλούς λόγους που θα αναφερθούν παρακάτω.
Μια διαμεμβρανική πρωτεΐνη αποτελείται, συνήθως, από ένα τμήμα "βυθισμένο" στην λιπιδική διπλοστοιβάδα - μεβράνη, και δύο άλλα τμήματα, ένα στον εξωκυττάριο χώρο, και ένα στον ενδοκυττάριο χώρο-κυτταρόπλασμα, στην γενική περίπτωση μιας πρωτεΐνης μεμβράνης κυτταρικής επιφάνειας (Σχήμα 1).
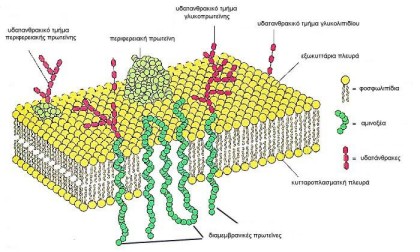
Η ακολουθία της μπορεί να "διαπερνάει" την λιπιδική διπλοστοιβάδα μία ή περισσότερες φορές, με άλλα λόγια μπορεί να έχει ένα ή περισσότερα διαμεμβρανικά τμήματα.
Ακόμη και με το υπέρ-αισιόδοξο σενάριο ότι στο άμεσο μέλλον ο πειραματικός προσδιορισμός της δομής των πρωτεϊνών θα αποτελεί υπόθεση ρουτίνας, για την τάξη των διαμεμβρανικών πρωτεϊνών δεν αναμένεται ριζική διαφοροποίηση από τα όσα μέχρι σήμερα ισχύουν. Είναι πολύ δύσκολο, για τη συντριπτική τους πλειοψηφία, να δώσουν μονοκρυστάλλους κατάλληλους για κρυσταλλογραφία ακτίνων-Χ (Persson and Argos, 1994; Aloy et al., 1997, Sussman et al., 1998), ενώ δίνουν συνήθως φτωχές πληροφορίες με τη φασματοσκοπία Πυρηνικού Μαγνητικού Συντονισμού (NMR). Επομένως, υπάρχουν πολύ λίγα δομικά δεδομένα, σε ατομική διακριτικότητα, ακόμη και στις μέρες μας. Τρισδιάστατες δομές σε υψηλή (ατομική) διακριτικότητα υπάρχουν για λίγες τέτοιες πρωτεΐνες (π.χ Photosynthetic Reaction Center, Βακτηριοροδοψίνη, Πορίνη) και σε χαμηλότερη διακριτικότητα (π.χ Plant light-harvesting complex II, Photosystem I, Nicotinic acetylocholine receptor).
Συνεπώς, για την κατηγορία αυτών των πρωτεϊνών οι προγνωστικές μέθοδοι είναι ακόμη πιο αναγκαίες από ότι για σφαιρικές-υδατοδιαλυτές πρωτεΐνες. Αν λάβουμε υπόψην τον περιορισμό στους βαθμούς ελευθερίας που επιβάλλουν τα διαμεμβρανικά τμήματα, γίνεται αυτονόητο πως στη συνέχεια η διαδικασία πρόγνωσης του διπλώματος είναι απλούστερη από ότι στις σφαιρικές πρωτεΐνες.
Μεγάλος αριθμός μεθόδων έχει σχεδιαστεί για τον προσδιορισμό της θέσης αλλά και της τοπολογίας διαμεμβρανικών τμημάτων σε μεμβρανικές πρωτεΐνες (von Heijne,1992; Persson and Argos, 1994; Cserzo et al., 1997). Οι πρώτες προσπάθειες στον τομέα αυτό, βασίζονταν σε υδροφοβική ανάλυση των ακολουθιών, για να εκμεταλλευτούν την προτίμηση που υπάρχει σε υδρόφοβα αμινοξικά κατάλοιπα μέσα στη λιπιδική διπλοστοιβάδα, εξαιτίας των ευνοϊκών αλληλεπιδράσεών τους με τις αλειφατικές περιοχές των λιπιδίων. Στην περίπτωση αυτή, ενώ οι περισσότερες διαμεμβρανικές περιοχές εντοπίζονται (von Heijne, 1992), έχουμε και αρκετές λανθασμένες προβλέψεις, οι οποίες αντιστοιχούν σε μη διαμεμβρανικά τμήματα με υψηλή υδροφοβικότητα.
Νεώτερες μέθοδοι, που βασίζονται σε πολλαπλές στοιχίσεις ομόλογων πρωτεϊνών δίνουν αισθητά βελτιωμένα αποτελέσματα, αποτυγχάνουν όμως όταν δεν βρίσκονται ομολογίες στις βάσεις δεδομένων. Στατιστικοί αλγόριθμοι, μπορούν να βοηθήσουν στην απόρριψη των ψευδών σημάνσεων, εντοπίζοντας πρότυπα που χαρακτηρίζουν τις διαμεμβρανικές περιοχές. Συνδυαστικοί αλγόριθμοι πρόγνωσης, δίνουν και εδώ αποτελέσματα με μεγαλύτερα ποσοστά αξιοπιστίας. Εξαιρετικό ενδιαφέρον έχουν επίσης αλγόριθμοι που αποσκοπούν στην πρόγνωση του προσανατολισμού των μη διαμεμβρανικών τμημάτων, στον ενδοκυττάριο ή εξωκυττάριο χώρο (για πρωτεΐνες της κυτταρικής μεμβράνης).
Σήμερα, η ανάγκη για καλύτερους και ταχύτερους αλγορίθμους πρόγνωσης, σε αυτόν τον τομέα, γίνεται επιτακτική, αν αναλογιστούμε τον τεράστιο όγκο πληροφοριών που διαρκώς συσσωρεύονται σε Βάσεις Δεδομένων πρωτεϊνικών ακολουθιών. Ενδεικτικά αναφέρουμε πως οι κατατεθειμένες στη SwissProt ακολουθίες διαμεμβρανικών πρωτεϊνών ήταν περίπου 2300 το 1994, ενώ σήμερα ξεπερνούν τις 10000. Το γεγονός ότι ο χρόνος που απαιτείται για να γίνει ανάλυση 'In Silico', με τη βοήθεια γρήγορων υπολογιστικών συστημάτων, είναι εξαιρετικά μικρότερος απ' αυτόν που θα απαιτούσαν οι πειραματικές μέθοδοι, στρέφει την προσοχή πολλών ερευνητών προς αυτήν ακριβώς την πλευρά.
Η έλλειψη δομικών δεδομένων για την κατηγορία των διαμεμβρανικών πρωτεϊνών μας αναγκάζει να υπολογίζουμε την αξιοπιστία των μεθόδων με βάσει τις καταχωρήσεις στην τράπεζα δεδομένων SWISS-PROT, παρόλο που υπάρχουν αρκετές διαφωνίες σε σχέση με την ακρίβεια των δεδομένων που αφορούν τις θέσεις των διαμεμβρανικών περιοχών (Cserzo et al., 1997). Κατά πλήρη αντιστοιχία με τις προγνώσεις δευτεροταγούς δομής η απόδοση των διαφόρων αλγορίθμων μπορεί να εκφραστεί ως εξής:
Ανά-κατάλοιπο(δύο καταστάσεων) : Ένα αμινοξικό κατάλοιπο, στην πρόγνωση που μας ενδιαφέρει, έχει δύο δυνατές καταστάσεις, δηλαδή να είναι διαμεμβρανικό (Τ) ή όχι (-). Τότε ανάλογα μπορούμε να υπολογίσουμε το δείκτη
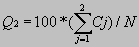 |
όπου Cj το πλήθος των καταλοίπων που προβλέφθηκαν σωστά σε μια δεδομένη κατάσταση j (T ή -) και Ν το συνολικό πλήθος καταλοίπων της ακολουθίας.
Ανά-διαμεμβρανικό τμήμα : Γίνεται με
Α) Μέτρηση του συνολικού πλήθους διαμεμβρανικών τμημάτων στην πρωτεΐνη.
Β) Υπολογισμό του μέσου μήκους τους,
και
Γ) Εύρεση της κατανομής του πλήθους διαμεμβρανικών τμημάτων σε συνάρτηση με το μήκος τους.
Η πρόγνωση της θέσης των διαμεμβρανικών τμημάτων γίνεται με αρκετά μεγάλη ακρίβεια. Υπάρχουν πολλές περιπτώσεις που επιτυγχάνονται αποτελέσματα με Q2 >> 90% . Βέβαια, οι μέσοι δείκτες αξιοπιστίας επηρεάζονται από την επιλογή του δειγματικού χώρου και φυσικά από τυχόν ανακρίβειες στις καταχωρήσεις της SWISS-PROT.
ΜΕΡΟΣ Α:
ΒΑΣΕΙΣ ΔΕΔΟΜΕΝΩΝ ΔΟΜΙΚΗΣ ΚΑΙ ΜΟΡΙΑΚΗΣ ΒΙΟΛΟΓΙΑΣ ΚΑΙ ΕΡΓΑΛΕΙΑ ΑΝΑΛΥΣΗΣ ΣΤΟ ΔΙΑΔΙΚΤΥΟ (INTERNET)
Στο πρώτο μέρος της άσκησης θα έχουμε πρόσβαση, μέσω του διαδικτύου, σε βάσεις δεδομένων, από τις οποίες θα έχουμε την ευκαιρία να 'αντλήσουμε' κάποιες πρωτεϊνικές ακολουθίες. Στη συνέχεια, θα επεξεργαστούμε την πρωτογενή αυτή πληροφορία με προγράμματα λογισμικού (εργαλεία ανάλυσης – analysis tools), που είναι επίσης ελεύθερα προσβάσιμα μέσω του Internet.
στην οποία είναι δυνατόν να επανερχόμαστε επιλέγοντας 'Home' ή 'Αρχική' σελίδα, ανάλογα με το φυλλομετρητή (Web-Browser).
C:\Sequences\[seq-name].seq
όπου '[seq-name]' το όνομα της ακολουθίας.
Ένα πρώτο βήμα στη μελέτη μιας 'άγνωστης' πρωτεΐνης είναι η αναζήτηση πιθανών ομολογιών μέσα στις βάσεις πρωτεϊνικών δεδομένων.
> Η ακολουθία πρέπει να εισαχθεί σε FASTA format.
> Είναι σημαντικό να γίνει κατανοητή η διαφορά των αποτελεσμάτων όταν έχει επιλεχθεί 'ungapped alignment'.
> Η προεπιλογή φίλτρου για περιοχές χαμηλής πολυπλοκότητας κάνει περισσότερο ευαίσθητο και αποδοτικό τον αλγόριθμο και καλό είναι να παραμείνει επιλεγμένο.
Ερώτηση:
Ποιο από τα προγράμματα που είναι διαθέσιμα πρέπει να χρησιμοποιήσουμε για να εντοπίσουμε ομολογία με άλλες πρωτεϊνικές ακολουθίες;
Παράδειγμα αποτελέσματος για την DYR_LACCA (Query) σε αντιπαραβολή με τον εαυτό της (Subject):
Score = 310 bits (785), Expect = 4e-84
Identities = 148/162 (91%), Positives = 148/162 (91%)
Query: 1 TAFLWAQDRDGLIGKDGHLPWHLPDDLHYFRAQTVGKIMVVGRRTYESFPKRPLPERTNV 60
TAFLWAQDRDGLIGKDGHLPWHLPDDLHYFRAQTVGKIMVVGRRTYESFPKRPLPERTNV
Sbjct: 2 TAFLWAQDRDGLIGKDGHLPWHLPDDLHYFRAQTVGKIMVVGRRTYESFPKRPLPERTNV 61
Query: 61 VLTHQEDYQAQGXXXXXXXXXXXXXXKQHLDQELVIAGGAQIFTAFKDDVDTLLVTRLAG 120
VLTHQEDYQAQG KQHLDQELVIAGGAQIFTAFKDDVDTLLVTRLAG
Sbjct: 62 VLTHQEDYQAQGAVVVHDVAAVFAYAKQHLDQELVIAGGAQIFTAFKDDVDTLLVTRLAG 121
Query: 121 SFEGDTKMIPLNWDDFTKVSSRTVEDTNPALTHTYEVWQKKA 162
SFEGDTKMIPLNWDDFTKVSSRTVEDTNPALTHTYEVWQKKA
Sbjct: 122 SFEGDTKMIPLNWDDFTKVSSRTVEDTNPALTHTYEVWQKKA 163
|
1 2 3 4 5 6
123456789012345678901234567890123456789012345678901234567890
. | . | . | . | . | . |
00000 HHHHHHHLLLLLLLLLLLLLLLLLLLEEEEEEEEEEEEEEEEEEELLLLLLLLLLLLEEE
00060 EEELLLLLLLLLEEHHHHHHHHHHHHHHHHHHHHEEEEEHHHHHHHHHHHHEEEEEEHHH
00120 HHHHHHHHLLLLLLLLLEELLLLLLLLLLLLLLLLLHHHHHH
|
|
|
ΣΥΝΤΟΜΗ ΠΕΡΙΓΡΑΦΗ |
|
|
Η πρώτη γραμμή ΚΑΘΕ καταχώρησης. Περιέχει τον Κωδικό PDB ID, μια βασική ταξινόμηση καθώς και την ημερομηνία κατάθεσης. |
|
|
Περιγραφή της Δομής που αναφέρεται στην καταχώρηση. |
|
|
Περιγραφή των 'Μακρομοριακών Περιεχομένων'. |
|
|
'Λέξεις Κλειδιά' που περιγράφουν το Μακρομόριο. |
|
|
Βιολογική προέλευση του μακρομοριόυ. |
|
|
Η πειραματική τεχνική που χρησιμοποιήθηκε για τη λύση της δομής. |
|
|
Κατάλογος των ερευνητών που εργάσθηκαν στον καθορισμό της δομής. |
|
|
Καθορισμός των θέσεων Υδρογονικών Δεσμών. |
|
|
Η Βιολογική Ακολουθία. Για πρωτεΐνες ακολουθείται ο κωδικός των τριών γραμμάτων (π.χ. ALA για Αλανίνη). |
|
|
Καθορισμός περιοχών με ελικοειδή δομή. |
|
|
Καθορισμός περιοχών με εκτεταμένη δομή. |
|
|
Παράμετροι Μοναδιαίας Κυψελίδας, Ομάδα Συμμετρίας Χώρου |
|
|
Πίνακας Μετατροπής από σύστημα ορθογωνίων συντεταγμένων στις συντεταγμένες που κατατέθηκαν αρχικά. |
|
|
Πίνακας Μετατροπής από σύστημα ορθογωνίων συντεταγμένων σε κρυσταλλογραφικές συντεταγμένες. |
|
|
Εγγραφές των ατομικών συντεταγμένων. |
|
|
Εγγραφές των ατομικών συντεταγμένων για τα Ετεροάτομα. |
|
|
Τερματισμός μιας Αλυσίδας. |
|
|
Τελευταία εγγραφή κάθε καταχώρησης. |
Τα αποτελέσματα ενός σημαντικού πειράματος δημοσιεύθηκαν στις αρχές του 1995: ερευνητές πρωτεϊνικών δομών, κατέθεσαν τις αμινοξικές ακολουθίες πρωτεϊνών (για τις οποίες ήταν έτοιμοι να έχουν σε σύντομο χρονικό διάστημα δομικά αποτελέσματα) δημιουργώντας κατ' αυτόν τον τρόπο μια 'Βάση-Δεδομένων-Προς-Πρόγνωση'.
Στη συνέχεια, για κάθε μια από τις καταχωρήσεις, ήταν δυνατόν να στείλει κάποιος την πρόγνωση της μεθόδου του πριν από κάποια καταληκτική ημερομηνία, η οποία συνέπιπτε με τη δημοσίευση της λυμένης δομής για το συγκεκριμένο μόριο. Τελικά, τα αποτελέσματα συγκρίθηκαν και συζητήθηκαν σε μια ειδική συνάντηση εργασίας (στο Asilomar της California), με τελικό συμπέρασμα ότι ακόμη δεν είναι δυνατή η πρόγνωση της δομής από την ακολουθία και μόνο.
Αυτό ήταν το γνωστό πείραμα CASP1, ενώ μετά από δύο χρόνια ακολούθησε και δεύτερο όμοιο (CASP2).
CASP1 (1995) Special issue of. Proteins.
CASP2 (1997) Special issue of. Proteins.
Aloy, P., Cedano, J., Olivia, B., Aviles, X. and Querol, E. (1997) 'TransMem': a neural network implemented in Excel spreadsheets for predicting transmembrane domains of proteins, CABIOS, 13(3), 231-234
Altschul, S. F., Gish, W., Miller, W., Myers, E. and Lipman, D. J. (1990) Basic Local Alignment Search Tool, J. Mol. Biol., 215, 403-410
Anfinsen, C. B. (1973). Principles that govern the folding of protein chains. Science, 181, 223-230.
Bairoch, A. and Apweiler, R. (1998) The SWISS-PROT protein sequence databank and its supplement TrEMBL in 1998, Nucleid Acids Res., 26, 38-42
Bairoch, A., Bucher, P., Hofmann, K.(1997) The PROSITE datatase, its status in 1997. Nucleic Acids Res. , 24, 217-221
Blout, E. R., de Loze, C., Bloom, S. M. and Fasman, G. D. (1960) Dependence of the conformation of synthetic polypeptides on amino acid composition, J. Am. Chem. Soc., 82, 3787-3789
Bork, P., Ouzounis, C. and Sander C. (1994) From genome sequences to protein function, Curr. Opin. Struct. Biol. , 4, 393-403
Chothia, C. and Lesk, A. M. (1986) The relation between the divergence of sequence and structure in proteins, EMBO J., 5 , 823-826
Chou, P. Y. and Fasman, G. D. (1978) Prediction of the secondary structure of proteins from their amino acid sequence, Adv. Enzymol., 47, 45-148.
Cserzo, K., Wallin, E., Simon, I., von Heijne, G. and Elofsson, A. (1997) Prediction of transmembrane a-helices in prokariotic membrane proteins: the dense alignment surface method, Protein Engineering, 10(6), 673-676
Doolittle, R. F. (1986) Of URFs and ORFs: a primer on how to analyze derived amino acid sequences, University Science Books, Mill Valley California.
Eisenberg, D., Weiss, R. M. and Terwilliger, T. C. (1984) The hydrophobic moment detects periodicity in protein hydrophobicity, Proc. Natl. Acad. Sc. U.S.A., 81, 140-144
Finkelstein, A. V. and Reva, B. A. (1992) Search for the stable state of a short chain in a molecular field, Prot.Engin., 5, 617-624
Gaasterland, T. (1997) Genome sequencing projects. Univ. Chicago, WWW document (http://www.mcs.anl.gov/home/gaasterl/genomes.html)
Garnier, J., Osguthorpe, D. J. and Robson, B. (1978) Analysis of the accuracy and Implications of simple methods for predicting the secondary structure of globular proteins, J. Mol. Biol., 120, 97-120.
Gibson, T. J., Thompson, J. D. and Abagyan, R. A. (1993) Proposed structure for the DNA-binding domain of the Helix-Loop-Helix family of eukaryotic gene regulatory proteins, Prot. Engin., 6 , 41-50.
Hamodrakas, S.J. (1988) A protein secondary structure prediction scheme for the IBM-PC and compatibles, Comput. Applic. Biosci., 4(4), 473-477
Holm, L. and Sander, C. (1996) The FSSP database: fold classification based on structure-structure alignment of proteins, Nucl. Acids Res., 24, 206-210
Hubbard, T. J. P., Murzin, A. G., Brenner, S. E. and Chothia, C. (1997). SCOP: a structural classification of proteins database, Nucl. Acids Res., 25, 236-239
Kabsch, W. and Sander, C. (1983a) How good are predictions of protein secondary structure?, FEBS Lett., 155,179-182
Kabsch, W. and Sander, C. (1983b) Dictionary of protein secondary structure: pattern recognition of hydrogen bonded and geometrical features, Biopolymers, 22, 2577-2637
Kyrpides, N. (1999) GOLD: Genomes On Line Database WWW-page (http://www.ebi.ac.uk/research/cgg/genomes.html)
Lemer, C. M.-R., Rooman, M. J. and Wodak, S. J. (1995) Protein structure prediction by threading methods: evaluation of current techniques, Proteins, 23, 337-355
Lesk, A. M. (1991) Protein Architecture - A Practical Approach, Oxford University Press, Oxford, New York, Tokyo.
Liakopoulos, T.D., and Hamodrakas, S.J. (1999) A new statistical mehod to predict the orientation of Non-transmembrane domains in transmembrane proteins, In preparation
Murzin, A.G., Brenner, S.E., Hubbard, T. and Chothia, C.(1995) Scop: a structural classification of proteins database for the investigation of sequences and structures, Journal of Molecular Biology, 247 , 536-540
Orengo, C. A., Michie, A. D., Jones, D. T., Swindells, M. B. and Thornton, J. M. (1997) CATH - A hierarchic classification of protein domain structures, Structure, 5(8) , 1093-1108
Palaios, G.A., and Hamodrakas, S.J. (1999) SecStr: Secondary Structure prediction over the World Wide Web, In preparation
Pasquier, C., Promponas, V.J., Palaios, G.A., Hamodrakas, J.S. and Hamodrakas, S.J. (1999) A novel method for predicting transmembrane segments in proteins based on a statistical analysis of the SwissProt database: the PRED-TMR algorithm, Protein Engineering, In Press
Pasquier, C. and Hamodrakas, S.J. (1999) An hierarchical artificial neural network system for the classification of transmembrane proteins, Protein Engineering, Submitted for publication
Persson, B. and Argos, P. (1994) Prediction of transmembrane segments in proteins utilising multiple sequence alignments, J. Mol. Biol., 237, 182-192
Piatigorsky, J and Wistow G. J. (1989) Enzyme/Crystalins: Gene Sharing as an Evolutionary Strategy, Cell , 57, 197-199
Rost, B. and Sander, C. (1996) Bridging the protein sequence-structure gap by structure predictions, Annu. Rev. Biophys. Biomol. Struct., 25, 113-136.
Rost, B. (1997) Protein structures sustain evolutionary drift, Folding and Design, 2, S19-S24.
Rost, B., O'Donoghue, S. and Sander, C. (1998) Midnight zone of protein structure Evolution, manuscript in preparation.
Rost, B. and Sander, C. (1993) Improved prediction of protein secondary structure by use of sequence profiles and neural networks, Proc. Natl. Acad. Sc. U.S.A., 90, 7558-7562
Scheraga, H. A. (1960) Structural studies of ribonuclease III. A model for the secondary and tertiary structure, J. Am. Chem. Soc., 82, 3847-3852.
Schulz, G. E., Baryy, C. D., Friedman, J., Chou, P. Y., Fasman, G. D. (1974) Comparsison of the predicted and observed secondary structure of T4 phage lysozyme, Nature, 250, 140-142
Sippl, M. J. (1982) On the Problem of Comparing Protein Structures. J. Mol. Biol., 156, 359-388
Sussman, J.L., Ling, D., Jiang, J., Manning, N.O., Prilusky, J., Ritter, O. and Abola, E.E. (1998) Protein Data Bank (PDB): Database of Three-Dimensional Structural Information of Biological Macromolecules, Acta Cryst. 54, 1078-1084
Szent-Gyorgyi, A. G. and Cohen, C. (1957) Role of proline in polypeptide chain configuration of proteins, Science, 126, 697.
Von Heijne, G. (1992) Membrane Protein Structure Prediction; Hydrophobicity Analysis and the Positive-inside Rule, J. Mol. Biol., 225, 487-494
Yona, G., Linial, N., Tishby, N. and Linial, M. (1998) A map of the protein space - An automatic hierarchical classification of all protein sequences, in the proceedings of ISMB 98, 212-221
Zu-Kang, F. and Sippl, M. J. (1996). Optimum superimposition of protein structures: ambiguities and implications. Folding and Design, 1, 123-132.
Diederichs, K., Freigang, J., Umhau. S., Zeth, K. and Breed, J. (1998) Prediction by a neural network of outer membrane b–strand protein topology, Protein Science, 7, 2413-2420
Fariselli, P and Casadio, R. (1998) HTP: a neural-network based method for predicting the topology of helical transmembrane domains in proteins, Comput. Applic. Biosci., 12(1), 41-48.
Hirokawa, T., Boon-Chieng, S. and Mitaku S. (1998) SOSUI: classification and secondary structure prediction system for membrane proteins, BioInformatics, 14(4), 378-379
Hobohm, U. and Sander, C. (1994) Enlarged representative set of protein strucures, Protein Science, 3, 522
Kihara, D., Shimizu, T. and Kanehista, M. (1998) Prediction of membrane proteins based on classification of transmembrane segments, Protein Engineering, 11(11), 961-970
Pasquier, C.M., Promponas, V.J., Varvayannis, N.J., and Hamodrakas, S.J. (1998) A Web server to locate periodicities in a sequence, Bioinformatics , 14(8) , 749-750.
Promponas, V.J., Palaios, G.A., Pasquier, C.M., Hamodrakas, J.S. and Hamodrakas, S.J. (1999) A Web Tool which combines transmembrane protein segment prediction methods, In Silico Biology, 3 , 1-4
Qian, N and Sejnowski, TJ. (1988) Predicting the secondary structure of globular proteins using neural network models, J. Mol. Biol., 262, 865-884
Reczko, M. (1993) Protein secondary structure prediction with partially recurrent neural networks, SAR and QSAR in Environmental Research, 1, 153-159
Rost, B., Casadio, R. Fariselli, P. and Sander, C. (1995) Transmembrane helices predicted at 95% accuracy, Prot. Sci., 4, 521-533
Rost, B., Fariselli, P. and Casadio, R. (1996) Topology prediction for helical transmembrane proteins at 86% accuracy, Prot. Sci., 5, 1704-1718
Rost, B. and O'Donoghue, S. I. (1997) Sisyphus and prediction of protein structure, CABIOS, 13, 345-356.
Rost, B., Sander, C. and Schneider, R. (1994) PHD – an automatic mail server for protein secondary structure prediction, CABIOS, 10(1), 53-60